空间定义的细胞间相互作用和串扰在解读驱动肿瘤发生和疾病进展的关键分子信息中非常重要。空间转录组、空间蛋白组学等多组学技术的发展极大推动了对复杂生物问题的理解。空间转录组学技术(Visium、DSP-WTA 等)虽能覆盖全转录组,但分辨率未达到单细胞水平。「进阶版」空间组学技术(Visium HD、Stereo-seq 等)在原有基础上结合生物信息学的细胞分割手段实现了单细胞分辨率。然而,深入解析细胞间精细的相互作用仍是一大难题。近期,Xenium、MERSCOPE 和 SMI 技术等在单细胞甚至亚细胞水平上实现了空间转录组的精准解析,为包括肿瘤微环境在内的多种疾病研究提供了新的的视角和工具(点击了解详情)。
非因生物作为国内第一家搭建 Xenium 空间单细胞转录组学技术的公司,率先利用该平台在乳腺癌样本中展开研究,并作为第一研究单位发表国内首篇 Xenium 应用预印版文章《Spatial Single-Cell Transcriptomic Analysis in BreastCancer Reveals Potential Biomarkers for PD-1Blockade Therapy》,该研究利用新开发的 Xenium 空间单细胞转录组学技术对四种具有不同生物学特征的乳腺癌(BC)组织样本(包括管腔型、HER2 2+ / HR-及三阴性乳腺癌,TNBC)进行了深入的空间单细胞分析,并将结果与传统病理学和预定义的兴趣区域(ROIs)进行了比较,以验证该方法的技术稳健性。
01 研究背景:
乳腺癌(BC)已成为应用空间转录组学和蛋白质组学技术研究的核心领域,尤其空间转录组学在研究乳腺癌肿瘤微环境(TME)方面发挥了至关重要的作用,涵盖了哺乳动物乳腺组织的空间解析表征及 HER2 阳性乳腺癌中多细胞结构的研究。并针对乳腺癌进行了初步研究,旨在探讨肿瘤内及肿瘤间的异质性。然而,尽管取得了这些进展,现有的转录组数据仍然不够全面,不足以阐明在不同类型的乳腺癌中单细胞水平上发生的复杂交互和串扰现象,亟需跟全面和深入的技术及分析方法。
02 研究流程:
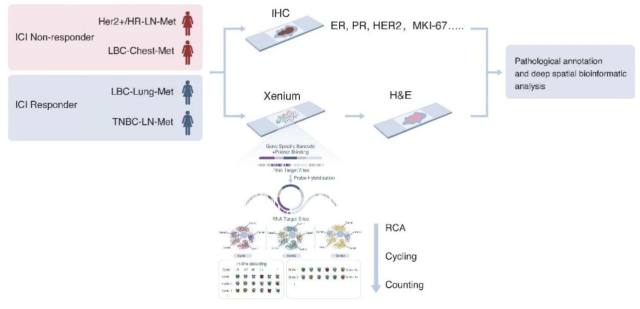
图 1 样本特征和实验流程示意图
03 研究结果:
1. 空间单细胞转录组学阐明不同乳腺癌类型中的独特细胞组成
本研究运用 Xenium 空间单细胞转录组学技术,针对胸壁转移的管腔癌(LBC-Chest-Met)、肺转移的管腔癌(LBC-Lung-Met)、HER2(2+)/HR-的癌伴淋巴结转移(Her2+/HR-LN-Met)以及淋巴结转移的三阴性癌(TNBC-LN-Met)四种不同病理特征的乳腺癌样本进行了深入分析,通过人乳腺组织的 280 基因探针 panel,获得了超过百万个细胞的转录组信息(图 2)。研究团队构建了一个多层级的细胞类型注释体系,涵盖了从主要细胞类别到具体免疫细胞亚群的详尽分类,并成功从所有样本中识别出 15 种不同的细胞亚型。样本间对比分析揭示了各类型乳腺癌中肿瘤微环境(TME)组成的显著差异,特别是 LBC-Lung-Met 中丰富的内皮和平滑肌细胞,反映了其独特的细胞环境。通过创建与 H&E 染色图像对应的细胞类型原位图,精确描绘了肿瘤及周围组织的结构,并发现了细胞类型间基于谱系的基因表达相关性,为理解乳腺癌的细胞异质性和 TME 提供了新的视角。
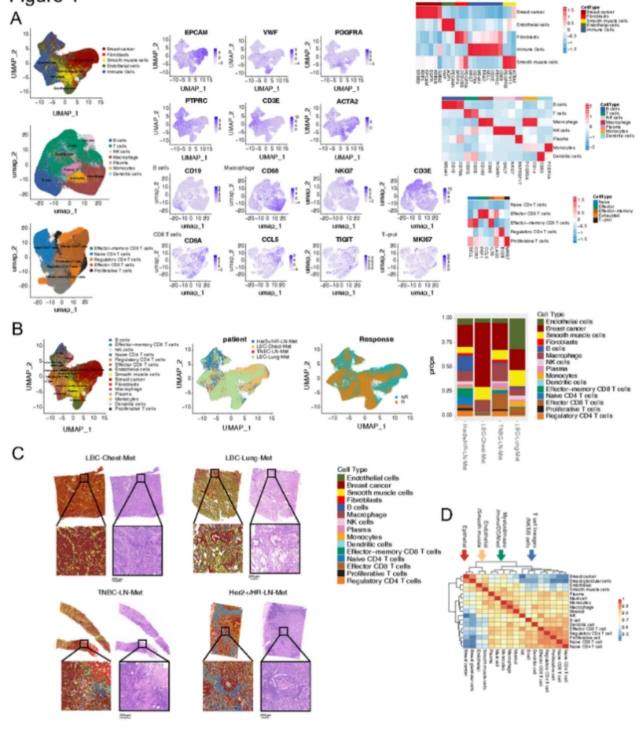
图 2 使用空间单细胞转录组学对乳腺癌组织中的细胞表型进行原位表征
2. 空间单细胞转录组数据的 IHC 验证
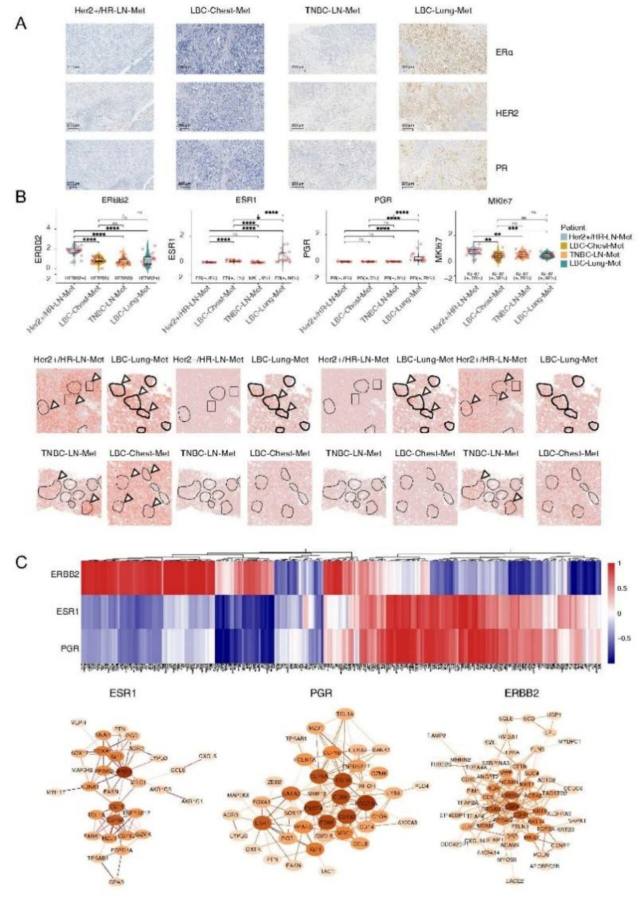
为评估 Xenium 空间单细胞数据的可靠性,研究团队将 mRNA 数据与来自相应样本的 IHC 定量蛋白数据进行了比较(图 3)。研究分析了四个关键蛋白标记物(ESR1、PGR、Her2、Ki-67)的表达,发现 LBC-lung-Met 样本中 ESR1 和 PGR 的蛋白表达显著,而 LBC-Chest-Met 中这两种蛋白表达极低,且 Her2 在 Her2+/HR-LN-Met 和 LBC-lung-Met 中呈 2+阳性,所有样本的 Ki-67 表达各异。为将 Seurat 标准化的单细胞 mRNA 水平与 IHC 数据进行比较,随机选取 20 个富含肿瘤的区域(ROI)进行详细比对,结果显示转录组数据与 IHC 蛋白表达情况高度吻合,比如 LBC-lung-Met 的 ERα 蛋白与 mRNA 高水平表达均得到验证,且 LBC-Chest-Met 的 mRNA 数据揭示了其与其他样本在 ERα 上的显著差异。此外,尽管某些情况下 mRNA 与蛋白水平不完全对应,如 LBC-Chest-Met 中的 PR 表达,总体上,Ki-67 的 mRNA 与蛋白一致性进一步证明了 Xenium mRNA 数据在定量分析中的准确性。随后,研究团队对 panel 中的所有基因进行了深入的空间单细胞 mRNA 特征分析,特别关注了基因间的相互关联,以此来识别与关键乳腺癌标志基因 ESR1、PGR 及 HER2 相关的调控网络。通过分层聚类分析,研究团队发现 ESR1 与 PGR 在调控网络中呈现紧密的协同作用,符合它们作为共轭激素受体的功能特点,而 HER2 则参与了一个独特的协同调控模式。利用 CytoSCAPE 软件进行网络分析进一步揭示,ESR1 和 PGR 不仅协同作用,还与激素受体共激活因子 PPARG 及转录因子 FOXA1、RUNX1 等紧密联系。这些激素受体不仅调控着激素信号传导,还参与到与 B 细胞和巨噬细胞的免疫调节中,凸显了它们在肿瘤微环境中的复杂角色。HER2 则构建了另一个包含 EGFR、角蛋白家族、CD9、PECAM 和 MKI67 等分子的独立信号网络。这些发现通过展示转录组学数据反映的调控网络多样性,强调了其在深入理解肿瘤生物学及微环境中的广泛应用潜力。
图 3 乳腺癌标志基因的 IHC 与 Xenium 空间单细胞转录组学数据对比分析
3. 空间单细胞转录组学揭示特定感兴趣区域内的独特细胞组成
在肿瘤微环境(TME)的特定区域中,研究团队进行了深入的调控网络分析,利用每个样本中的 20 个肿瘤区域兴趣点(ROIs),这些区域的 mRNA 表达与组织学特征相符。另外,从三个样本中各自挑选 10 个免疫-基质 ROIs 进行对比,但 LBC-lung-Met 未纳入分析,因为其缺乏明确的免疫-基质区域(图 4)。研究发现,肿瘤富集的 ROIs 显示了多样的 TME 结构,LBC-Chest-Met 的肿瘤细胞比例最高(78.5%),而其他转移样本的肿瘤细胞比例较低(37.2%-63%),伴随大量淋巴细胞浸润。与肿瘤 ROIs 相比,免疫-基质 ROIs 揭示了非肿瘤细胞的多样性,LBC-Chest-Met 与 TNBC-LN-Met 在细胞组成上相似,但特定细胞类型分布有显著差异。尽管免疫细胞浸润程度不同,但是所有样本均表现炎症性 TME。利用邻近度指标分析肿瘤 ROI 中的细胞共定位,研究团队观察到每个乳腺癌样本独特的细胞共定位模式。通过空间转录组学分析,研究团队清晰地描绘了每种乳腺癌亚型中 TME 的特有复杂结构,特别是在高肿瘤含量区域。
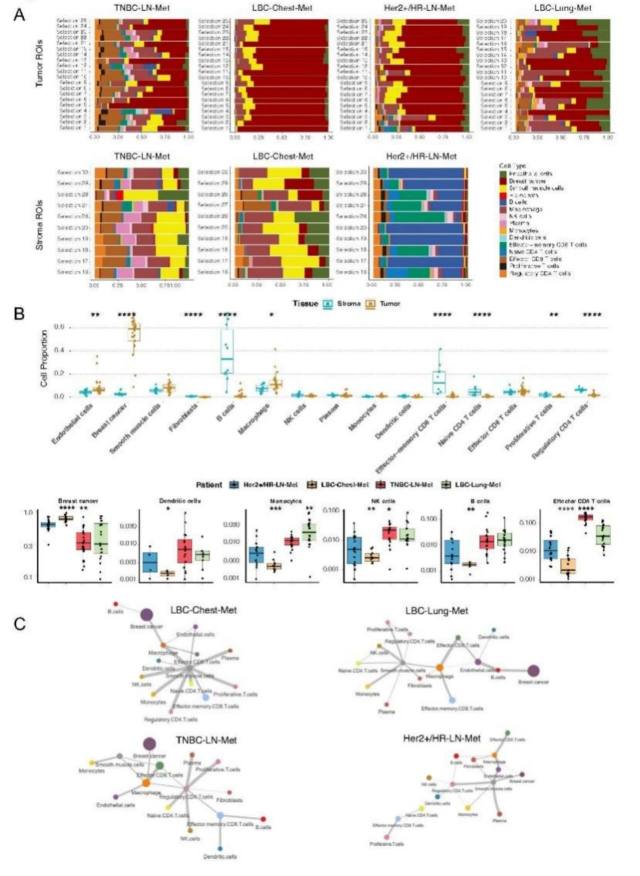
图 4 乳腺癌肿瘤微环境中的细胞组成与通讯
4. 空间单细胞转录组学揭示免疫细胞定位及其治疗意义
随着乳腺癌 TME 空间单细胞转录组图谱的建立,研究团队旨在探究与抗 PD-1 疗法临床反应相关的细胞网络。首先比较了响应者(R:TNBC-LN-Met 和 LBC-Lung-Met)与非响应者(NR:LBC-Chest-Met 和 Her2+/HR-LN-Met)的肿瘤 ROIs(图 5)。研究发现,相比于非响应者,响应者肿瘤中 T 细胞亚群及免疫细胞的比例增加,表明这些免疫成分具有潜在的抗肿瘤作用。通过构建细胞邻域图谱,鉴定出十种不同的细胞集群(CN1-CN10),并指出在响应者中,富含效应 T 细胞的 CN7 和包含肿瘤交互巨噬细胞及效应 T 细胞的 CN5 集群更为显著,提示这些细胞群在抗 PD-1 治疗响应中扮演关键角色。此外,研究团队计算了每个细胞在 100μm 半径内与肿瘤的距离。在一致的细胞毒性 T 细胞中,在 CN5 中表现出肿瘤募集模式,通常比非响应者更靠近肿瘤。同样,与肿瘤相互作用的巨噬细胞表现出与响应者中肿瘤细胞更紧密的定位。值得注意的是,在 CN5 中,以 Ki-67 表达为标志的增殖性 T 细胞亚群,但在响应者的肿瘤邻近生态位中明显更为普遍,而不是非响应者,强调了它们在 T 细胞介导的肿瘤细胞杀伤中的作用。研究团队进一步研究了所有细胞类型在肿瘤富集区与整个组织相比的空间富集评分(ESscore)。在响应者中,观察到效应 T 细胞、增殖性 T 细胞和巨噬细胞的显着空间共募集现象与先前研究及 Ki-67 的 IHC 结果相吻合。总而言之,这些空间分析验证了一个强大的框架,该框架将临床表型与空间单细胞转录组数据相结合,这种方法有望通过更多样本获得进一步的见解。
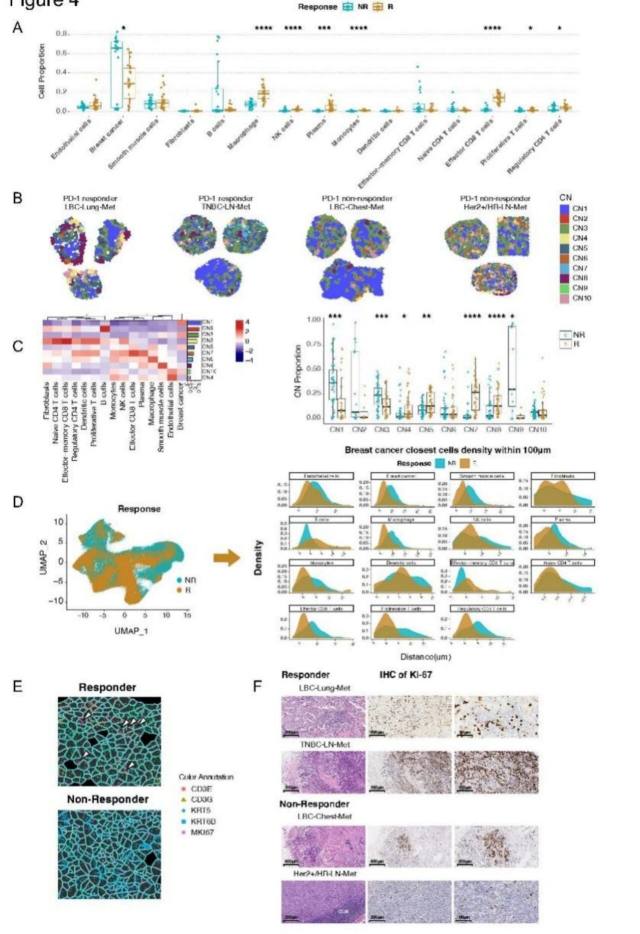
图 5 肿瘤区域(ROI)中细胞邻域(CN)和单细胞水平的独特细胞组织结构
5. 细胞通讯揭示了空间定义的配体/受体对作为潜在的生物标志物
研究通过 CellphoneDB 分析了肿瘤微环境(TME)中细胞间通讯,所有样本中均发现巨噬细胞通过抑制性 CD86/CTLA4 相互作用与 T 细胞通讯。相反,仅在响应者中,巨噬细胞表达的 CD274(PD-L1)与 DCs、T 细胞(增殖性 T 细胞和 CD4+调节性 T 细胞)及巨噬细胞自身表面的 CD80 显示出显著的交互作用。表明在响应者 TME 中存在一种 T 细胞激活细胞通讯模式。此外,除了巨噬细胞,效应 T 细胞亚群在响应者中也独有这种抑制性 CD86/CTLA4 交互,这可能是广泛激活 CD8+ T 细胞的结果。值得注意的是,刺激性的 CD274/CD80 交互也在一名响应者(TNBC-LN-Met)的 CD8+ T 细胞和其他免疫细胞之间被观察到。并且,CD274/CD80 的交互在响应者的肿瘤细胞与免疫细胞间被发现。与之相符,Xenium 数据的空间细胞注释显示,仅在响应者中,CTLA4 阳性的效应 T 细胞和 PD-L1 阳性的巨噬细胞直接与癌细胞通讯。
鉴于 PD-L1 作为 ICI 疗法反应生物标志物的既定作用,研究团队检测了样本肿瘤细胞中 CD274 及其他基因的 mRNA 水平。肿瘤细胞中与免疫激活相关的基因,尤其是 PDCD1LG2、NKG7 和 CD80 的 mRNA 水平在响应者中显著提升,表明它们可能是优于 PD-L1 的潜在生物标志物。这进一步证实了研究结果的有效性,并预示了在空间解析的组织背景下发现生物标志物的潜力。
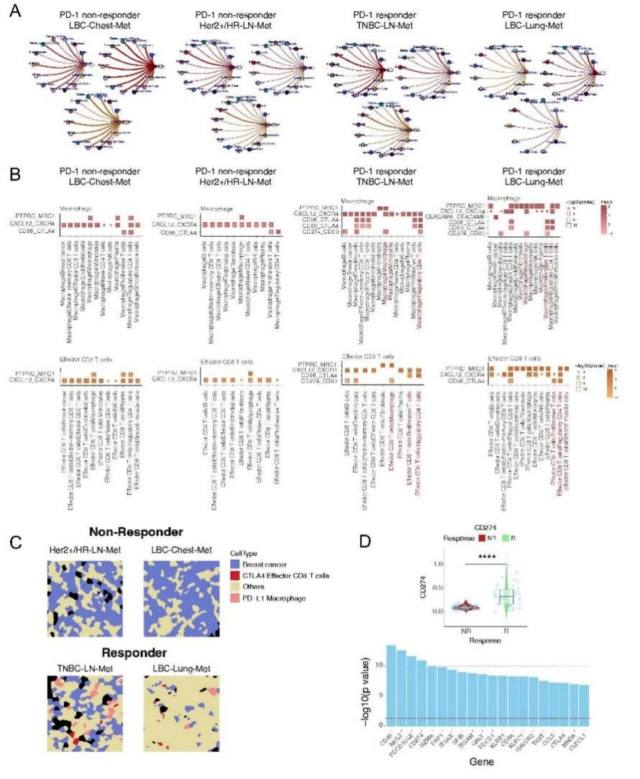
图 6 细胞通讯分析揭示了巨噬细胞-T 细胞配体-受体相互作用可作为潜在的生物标志物
6. 整合 scRNA-seq 和 Xenium 数据的基因推断
为了增强空间单细胞转录组分析的深度,研究团队将 scRNA-seq 数据与 Xenium 数据进行整合,运用与 Xenium 兼容的 SpaGE 和 Tangram 两种算法进行基因表达推断。通过将 Xenium 数据的三分之二作为训练集,研究团队训练模型预测 scRNA-seq 中所有基因的空间表达模式,并以剩余三分之一为验证集,验证了预测准确性。尽管低表达基因预测较为困难,但这两算法在中高表达基因上展现了可靠的空间表达映射能力。总之,这些推断算法成功扩展了我们对基因表达空间分布的理解,补全了 Xenium 数据集之外的信息,促进了更广泛基因表达图谱的绘制。
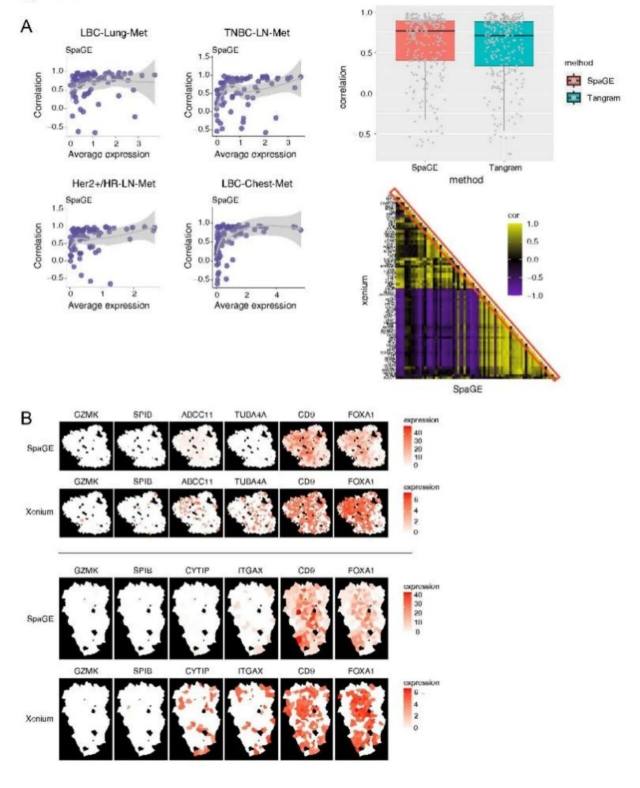
图 7 使用 SpaGE 和 Tangram 进行空间基因推断
04 结论
本研究对四个具有不同生物学特征的乳腺癌(BC)组织样本进行深入的空间单细胞剖析,通过选择预定义的兴趣区域(ROIs),研究团队巩固了该方法在转录水平上界定不同分子亚型的技术稳健性,使之与经典的免疫组化结果相匹配。此外,还证明了高维度的空间转录组(ST)数据能够识别出一个主要的细胞网络,该网络通过肿瘤邻近细胞区域内巨噬细胞与细胞毒性 T 细胞之间的相互作用,经由 PD-L1/CD80 和 CD86/CTLA4 轴相互交织,这一现象反映了临床观察到的 PD-1 介导的药物反应改善。通过整合开源计算方法(Tangram 和 SpaGE),研究团队找到了与之兼容的原位表达推断工具,这一方法具有通用性,能够利用 Xenium 原位或其他并行技术实现更深层次的空间剖析。