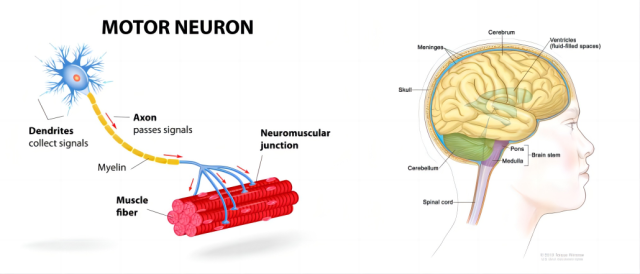
概述
肯尼迪病又称脊髓延髓肌萎缩症 (Spinal bulbar muscular atrophy,SBMA) 是一种 X 连锁隐性 (OMIM:313200,XLR) 是一种控制肌肉运动的特殊神经细胞(运动神经元)的疾病。这些神经细胞起源于脊髓和大脑中与脊髓相连的部分。患者表现为不同程度的下运动神经元损害、感觉障碍及内分泌系统异常,包括男性乳房发育、睾丸萎缩、轻度不育以及糖尿病等。
病因和流行病学
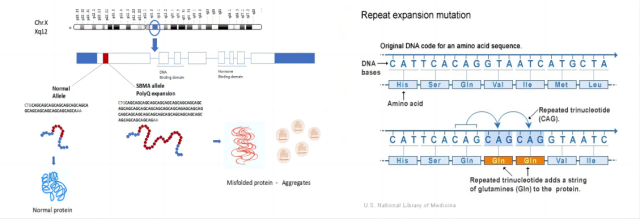
SBMA 是由染色体 Xq11-12 上的雄激素受体 (androgen receptor,AR) 基因第 1 号外显子 CAG 重复序列异常扩增所致。研究人员认为,含有 CAG 片段的雄激素受体蛋白片段在这些细胞内积累,并干扰正常细胞功能。神经细胞逐渐死亡,导致这种情况下的肌肉无力和萎缩。CAG 重复次数较多的人往往在更早的年龄出现脊髓和延髓肌萎缩的体征和症状。
SBMA 主要在成年男性中发病,女性携带者一般无明显症状。在美国,肯尼迪病的发生率为男性中 1/40 000,但芬兰西部和意大利报道的发病率更高。尚缺乏肯尼迪病在中国人群中发病率等流行病学数据。
临床表现
男性发病,一般起病隐匿,常见发病年龄为 30-60 岁。患者常以痛性痉挛、震颤、双下肢无力等症状起病,逐渐出现双上肢无力、延髓部和面部的肌肉萎缩、肌束颤动、构音障碍、吞咽困难、呼吸困难等。可伴有男性乳房发育、生殖功能降低等雄激素受体不敏感表现。约超过 50% 的患者存在感觉异常。部分女性基因突变携带者有轻度临床症状,可仅出现痉挛,电生理检查所示慢性失神经改变多轻微。该病进展缓慢,患者通常在病程晚期才出现行走不能,仅有部分患者需要辅助通气,对生存期无显著影响。
01|不典型症状
某些不典型症状可在出现肢体力弱前数年甚至数十年前出现,包括容易疲劳、痛性痉挛、口周震颤及姿势性震颤。肌无力:多数患者以双下肢无力起病,逐渐进展至双上肢、舌肌以及面肌,而眼外肌往往不受累。肢体无力的分布多不对称,以近端受累为主。咀嚼肌受累时可引起下颌下垂及震颤。体检以下运动神经元损害体征为主,表现为肢体肌力下降,近端为著,伴肌肉萎缩、束颤,腱反射多减弱或消失。多数患者可出现明显舌肌萎缩、纤颤,但软腭活动、咽反射多正常,患者饮水呛咳、吞咽困难的程度也较轻。某些患者还可观察到颏部以及口周束颤。患者运动功能损害随病程延长逐渐加重,后期可出现上楼费力、行走不稳等。
02|感觉异常
约半数患者存在不同程度的感觉减退,而在其他患者为亚临床性,仅在感觉神经传导检查中发现异常。
03|内分泌异常
包括葡萄糖及脂肪代谢的异常,雄激素受体不敏感等表现,包括男性乳房发育、性功能下降、不孕不育、睾丸萎缩等。
04|女性携带者
多数女性携带者并不出现症状,一般出现症状程度较轻,可仅表现为束颤、轻度远端肢体无力、肌肉痉挛或肌酸肌酶增高等。
辅助检查
1. 常规化验:肯尼迪病患者血清肌酸肌酶和乳酸脱氢酶可轻度或明显升高,性激素包括睾酮、促卵泡激素、黄体生成素水平也可出现异常。腰穿脑脊液检查通常正常。某些患者可出现高脂血症以及糖耐量受损。
2. 电生理检查:神经传导检查可提示感觉神经动作电位波幅降低,感觉神经传导速度减慢。针极肌电图多呈广泛神经源性损害,存在进行性和(或)慢性失神经改变,出现多个自发电位,运动单位动作电位时限显著增宽,甚至出现巨大电位,大力收缩时呈单纯相。单纤维肌电图上 jitter 明显增宽,运动单位计数也明显减少。
3. 肌肉活检:主要表现为神经源性损害,有时可合并肌源性损害特征。
4. 神经活检:腓肠神经活检可见大的有髓纤维减少,少量纤维脱髓鞘,施万细胞变性。
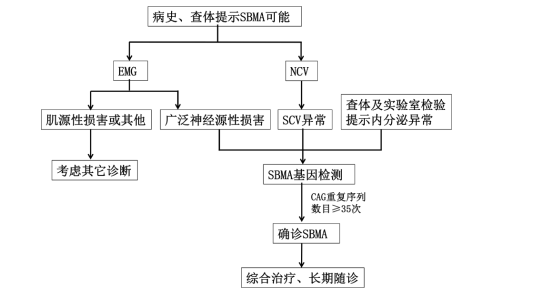
5. 基因检测:不同文献来源,CAG 次数略微有差异。既往多数文献把 CAG 拷贝次数大于 40 次作为确诊标准。2011 年欧洲神经科学联合会指南将 CAG 重复序列数目 ≥ 35 次作为诊断 SBMA 的依据。一般 CAG 重复 36-37 次等位基因临床意义的解释应该结合家族历史,以及该先证者的临床表现与其他家庭成员的基因型与表型的相关性进行综合判定。CAG 异常扩增长度与发病年龄和起病症状有关,与疾病的进展无关。与其他突变基因重复扩增的疾病相似,SBMA 亦呈现「遗传早现」现象,重复拷贝数在传代过程中不断增加,导致发病时间逐代提前,症状逐代加重。
诊断
AR 基因中 CAG 重复序列扩增数目是诊断肯尼迪病的金标准。根据病史、临床检查及神经电生理表现,结合基因检测结果即可明确诊断。
鉴别诊断
肌萎缩侧索硬化(ALS)家族性 ALS 约占患者总数的 5%-10%,多为常染色体显性遗传,极少数为 X 连锁遗传。患者同时存在不同程度的上下运动神经元受累表现。ALS 病情较 SBMA 进展快,最终多因呼吸肌麻痹或并发呼吸道感染死亡,生存期通常 3~5 年。而 SBMA 只累及下运动神经元,呈对称性缓慢进展,预后通常比较良好,预期寿命几乎不受影响或仅有小幅缩短。此外,ALS 无雄激素不敏感表现,结合基因检测、电生理、生化、影像学方法可鉴别。
进行性肌萎缩(PMA)PMA 是一种进行性下运动神经元的疾病,被认为是运动神经元病的一种,与典型 ALS 相比生存期可能更长。多以单侧肢体远端肌无力、肌萎缩起病,并逐渐向近端进展。与 SBMA 不同点在于,肌电图检查可见远端 CMAP 波幅明显下降,感觉神经传导测定通常为正常。SBMA 由于较多累及近端肌肉,其远端 CMAP 多处于正常范围,而感觉神经传导测定提示感觉神经动作电位波幅降低,感觉神经传导度减慢。
成人型脊髓性肌萎缩症(SMA)成人型 SMA 以常染色体隐性遗传为主,病变主要累及脊髓前角运动神经元,以四肢近端肌无力、肌萎缩为主要表现。但前者女性也可发病,较少呈 X 连锁隐性遗传,且无男性乳房发育等内分泌改变和感觉受累的症状,基因检测仍然是鉴别诊断的关键。
其他其他疾病包括免疫介导性(如重症肌无力、多灶性运动神经病、多发性肌炎),内分泌相关性(如甲状腺功能亢进),遗传性疾病(如肌营养不良、线粒体疾病)等引起的肢体无力,均应与 SBMA 相鉴别。遗传咨询女性突变携带者通常不会在临床上表现出来,但有 50% 的风险将突变遗传给后代。受累的男性一般不会遗传此疾病,有生育能力的受影响男性会遗传给每个女儿,他们的女儿 100% 可能成为突变携带者。
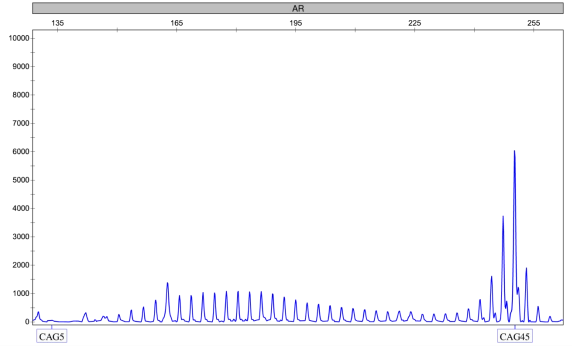
案例分享:
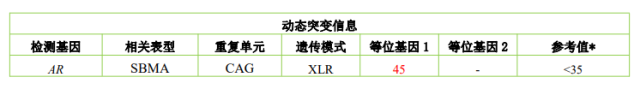
临床信息:男,38 岁,肯尼迪病
检测项目:AR 基因动态突变检测
检测结果:AR 基因为 CAG45 次重复
参考:
1.Breza M, Koutsis G. Kennedy's disease (spinal and bulbar muscular atrophy): a clinically oriented review of a rare disease. J Neurol. 2019 Mar;266(3):565-573. doi: 10.1007/s00415-018-8968-7. Epub 2018 Jul 13. PMID: 30006721.
2.Grunseich C, Fischbeck KH. Molecular pathogenesis of spinal bulbar muscular atrophy (Kennedy's disease) and avenues for treatment. Curr Opin Neurol. 2020 Oct;33(5):629-634. doi: 10.1097/WCO.0000000000000856. PMID: 32773451; PMCID: PMC7748295.
3.GeneReviews( ncbi.nlm.nih.gov/books/NBK1333/)
4.《罕见病诊疗指南 2019 年版》
5.orpha.net/
6.medlineplus.gov/