随着科技飞速发展,人工智能(AI)技术已经渗透到了各个领域,为人类带来了前所未有的便利。在生命健康领域,AI 正以其独特的能力,在基因组医学的创新和发展中发挥着关键作用。特别是在罕见遗传病的诊断上,AI 展现出了巨大的潜力。目前全球有 2.63 ~ 4.46 亿罕见遗传病人群,其中约半数的罕见病疑似病例未得到诊断,而已确诊病例的平均诊断周期约为 5 ~ 6 年,最长的甚至要等待数十年。国际罕见病联盟的第二个十年计划(2017-2027)的首要目标就是「让所有罕见病患者在就诊后的一年内获得精准的诊断」。基因测序技术的临床应用,可以使一些分子机制已知的罕见病的检测周期缩短至数周。然而要大规模、快速而准确地从海量的基因组变异中识别出致病因素,仍然是一个挑战。以基因检测大数据为基础,AI 算法为核心的智能化筛选工具的开发,正在加速相关罕见病的诊断进程。
AI 在罕见遗传病筛选致病变异方面的进展
在过去的十年里,人工智能(AI)在罕见遗传病的致病基因和变异筛选方面取得了显著进展。这些方法主要利用语义相似性关联患者的表型与已知的致病基因,结合变异位点的注释信息构建评分或预测模型,实现对候选变异的过滤及排序,代表性工具包括: Exomiser[1], VAAST[2], Xrare[3], AMELIE[4], GEM[5], Emedgene[6], and AI-MARRVEL[7]等,其中 AI-MARRVEL(AIM)是今年 4 月新英格兰医学 AI 杂志发表的新方法,研究人员基于随机森林算法对内部数千个确诊病例的超 350 万个变异进行训练,可根据患者的临床特征和测序数据对孟德尔遗传病的潜在致病基因或变异进行优先排序,显著提高遗传病诊断的准确率。
尽管上述工具在临床诊断中展现了很高的应用价值,但它们大多数基于表型语义相似性度量,这要求将患者的临床表型信息转化为标准化的表型词条,例如人类表型本体(HPO)。这一转化过程既是知识密集型的工作、又是劳动密集型的工作,往往需要遗传分析专家的校准,耗时耗力的同时,也高度依赖于基因型与表型关联数据库的全面性和患者临床表型描述的精准度。
大语言模型(large language model,LLM)的最新进展,为罕见病的分析解读提供全新的思路。如近期沙特阿拉伯阿卜杜拉国王科技大学和美国费城儿童医院的两个研究团队先后评估了大型语言模型在罕见遗传病诊断中进行基因优先排序的实用性。美国费城儿童医院的研究团队的结果表明尽管目前 LLMs 在生成准确的候选基因预测结果方面落后于传统工具,但随着模型规模的增加,它的性能有望进一步提高,尤其在处理非结构化文本数据时,LLM 展现了一定的优势[8]。而沙特阿拉伯阿卜杜拉国王科技大学的研究团队则通过真实的临床数据研究,展示了 LLMs 在基于表型的基因优先级排序任务中,不仅能够提供和传统工具相媲美甚至略优的基因排名,还能生成解释性的结果,有助于更高效的揭示基因与疾病之间的复杂联系[9]。上述这些研究结果表明,LLMs 凭借其在处理大规模文本数据和复杂问题上的能力,有望辅助临床医生和研究人员更高效地识别疾病相关的基因和变异,从而推动个性化精准医疗的飞速发展。
华大基因基于大语言模型的新方法带来新范式
作为全球精准医学和公共卫生领域的引导者,华大基因长期专注于以多组学大数据技术助力科研与临床应用转化、推动生命科学研究进展、生命大数据应用和提高全球医疗健康水平。为了持续提高遗传病分析解读的水平,华大基因 AI 团队在近期的研究工作中首次应用微调大语言模型来识别罕见遗传疾病的致病变异,开发了大语言模型驱动的新方法 Genetic Transformer(GeneT),相关工作在 medRxiv 预印[10]。该方法利用了公开数据构建的数万例阴阳性病例作为训练数据集,将基于资深遗传病分析专家们解读思维链构建的提示词作为模型微调的逻辑基础,引导基础大语言模型学习罕见遗传病致病变异筛选的能力。GeneT 在模拟样本和真实临床样本中分别达到 99% 和 98% 的致病变异召回率,同时分析效率提升了 20 倍。这一成果有望替代传统表型驱动的筛选方法,促进罕见遗传病的研究和临床应用,帮助广大患者群体获得精准诊断和治疗。
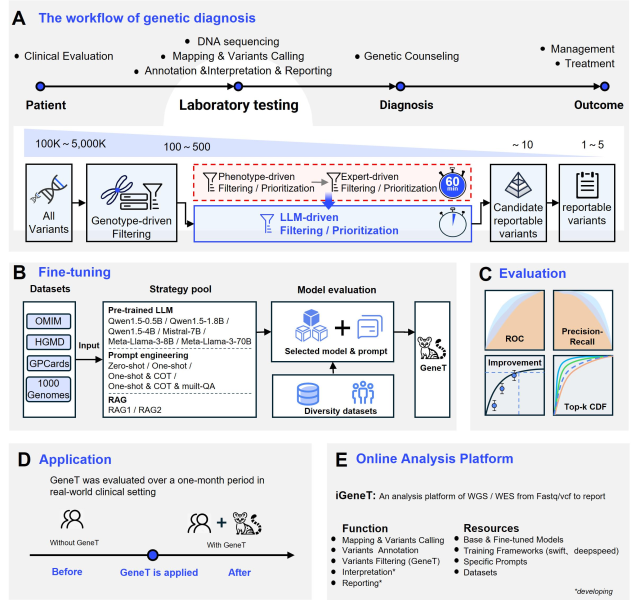
图 1:GeneT 研究概览
研究内容
一、预训练模型的选择和特征评估
我们利用公开数据构建的数万例阴阳性病例以及解读专家精心设计的问答提示词,构建了一组不同数据量梯度的训练数据。随后,我们对 6 个不同参数量的大语言模型进行了微调,以使模型专注于致病变异筛选这一任务。结果表明:
在较少训练集(n = 800)时,微调模型已经展现出了不错的预测性能,达到了 90% 的 F1 分数。随着数据量的增加,模型准确性稳步提升,当训练集拓展至 20000 时,F1 超过 99%。
小参数量的 LLMs,如 Qwen-1.5-0.5B 和 Qwen-1.5-1.8B,具有和大参数量的 LLMs 相当甚至更优的最佳预测性能。考虑小参数量 LLMs 部署所需硬件配置要求更低、内存占用更小、响应时间更快,在时效要求高、资源有限、注重隐私和安全的场景下将会是更优的选择。
通过「特征递增实验」,我们发现使用变异基础信息加上变异所在基因相关的疾病特征时,模型预测效果得到了显著增强,这强调了基因疾病信息在模型判定变异致病性中的关键作用,与实际解读人员的变异筛选经验相符。
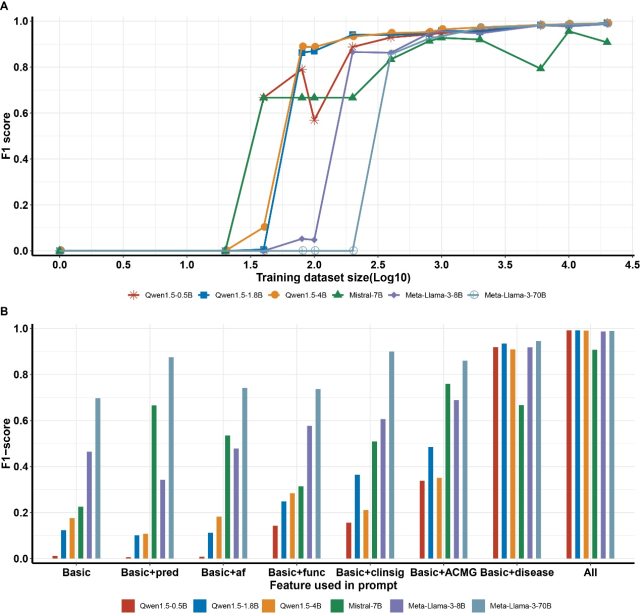
图 2:大语言模型在不同数据量梯度下微调后的性能表现及特征评估
二、多元化表型适用性和性能评估
我们使用来自 HGMD,OMIM,GPCards 三个数据库的已知致病变异和千人基因组的变异分别构建了 200 个模拟样本,用于评估模型的性能。这三个数据库的病例的表型形式不一,HGMD 样本的表型以疾病名为主,OMIM 样本的表型为自由文本形式的描述,而 GPCards 样本的表型为标准的表型词条。测评结果表明:
GeneT 显著降低了候选变异数目
GeneT 在三种表型测试集上效果都显著优于现有最优排序方法
GeneT 使用预测概率作为置信度分值,对所有候选位点排序,可替代现有的排序工具
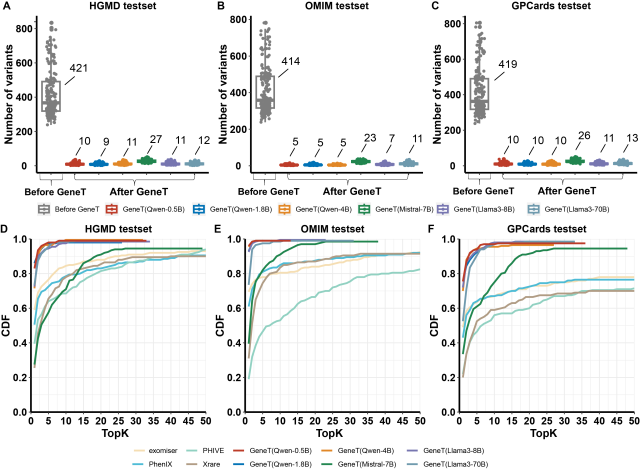
图 3:GeneT 在不同表型数据集上的表现以及和现有工具的性能对比
三、真实临床场景的应用评估
GeneT 方法在真实的临床解读场景中进行了应用,比较了应用前后一个月的时间消耗和召回率情况。为保证结果的可靠性,一般会安排专家 1 和专家 2 分别进行候选报出变异的初步筛选和初筛结果复核。
应用了 GeneT 辅助的专家 1 在选点环节的时间消耗及召回率情况评测结果如下:
1. 更快的分析时间:
参考模型推荐位点进行初筛时,专家 1 能更快定位阳性位点,观察到耗时从 60 分钟降低到约 44 分钟,有显著提升。
若初筛环节直接使用 GeneT 结果,时间可以从 60 分钟缩短到约 3 分钟,实现约 20 倍的效率提升。
2. 更准的选点:初筛环节无论仅参考还是直接使用模型选点结果,均能在召回率方面有所提升,从原来的 94.36% 上升到 97.40%/97.85%。
3. 更稳定的个体表现:因专家解读经验和样本解读难度差异,应用 GeneT 之前各解读专家之间约有 20% 的召回率差距。模型应用后解读专家在召回率表型上差异明显缩小到 5%,趋近于大语言模型工具的表现。
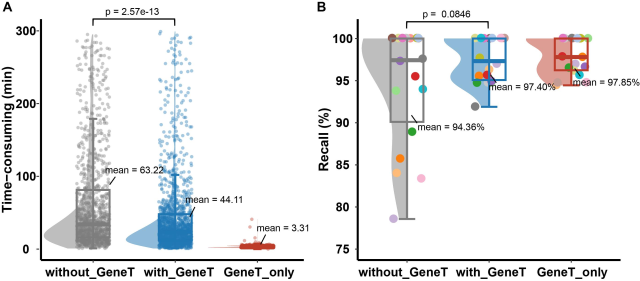
图 4:真实临床场景下, GeneT 的分析效率和性能表现
结语
华大基因 AI 研究团队开创性引入了大语言模型来识别罕见遗传疾病的致病变异,并利用真实临床场景验证展示了其相较于传统工具及遗传病人工筛选的优势,可以更灵活的面向多样化的临床表型信息、更高效精准的进行变异筛选及排序、结合 RAG 技术还可以实现更新更及时的数据库检索功能等,有望解决现有遗传病分析解读的痛点和难点。
这项研究成果代表了一种遗传病解读分析方法的范式转变,除了应用在候选致病变异的筛选环节,在基于表型的疾病识别、变异解读分类、基因组咨询等多个环节,都展露出了大语言模型具备的强大应用潜力。华大基因 AI 研发团队将持续深耕大语言模型在相关领域的研究与应用,助力精准医学的发展。
参考文献:
[1] Smedley, D., et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat. Protoc. 2015;10(12):2004-2015.
[2] Flygare, S., et al. The VAAST Variant Prioritizer (VVP): ultrafast, easy to use whole genome variant prioritization tool. BMC Bioinform. 2018;19(1):57.
[3] Li, Q., et al. Xrare: a machine learning method jointly modeling phenotypes and genetic evidence for rare disease diagnosis. Genet Med 2019;21(9):2126-2134.
[4] Birgmeier, J., et al. AMELIE speeds Mendelian diagnosis by matching patient phenotype and genotype to primary literature. Sci Transl Med 2020;12(544).
[5] Vega, F.M.D.L., et al. Artificial intelligence enables comprehensive genome interpretation and nomination of candidate diagnoses for rare genetic diseases. Genome Medicine 2021;13(1):153.
[6] Meng, L., et al. Evaluation of an automated genome interpretation model for rare disease routinely used in a clinical genetic laboratory. Genet Med 2023;25(6):100830.
[7] Mao, D., et al. AI-MARRVEL — A Knowledge-Driven AI System for Diagnosing Mendelian Disorders. NEJM AI 2024;1(5).
[8] Kim, J., et al.. Assessing the Utility of Large Language Models for Phenotype-Driven Gene Prioritization in Rare Genetic Disorder Diagnosis. arXiv preprint arXiv:2403.14801.
[9] Kafkas, Ş., et al. The application of Large Language Models to the phenotype-based prioritization of causative genes in rare disease patients. medRxiv 2023:2023.2011.2016.23298615
[10] Liang, L., et al. Genetic Transformer: An Innovative Large Language Model Driven Approach for Rapid and Accurate Identification of Causative Variants in Rare Genetic Diseases. medRxiv 2024:2024.2007.2018.24310666.