【流行病学】
一过性骨髓增生异常髓系白血病(Transient abnormal myelopoiesis TAM associated with down syndrome,)是一种罕见的类型白血病,约 10% 患有唐氏综合征的新生儿出现一过性的骨髓异常造血,多数病例数周至 3 月内自行缓解;临床 TAM 最常见于新生儿早期。就诊时的中位年龄为 3-7 天;大多数病例会在 2 个月内出现,但最多可在 6 个月大时确诊。
唐氏综合征患儿白血病发病率较正常儿童高 50 倍,至少半数为髓系,其中大部分为急性巨核细胞白血病 (Acute megakaryocytic leukemia ,AMKL)[1]。AMKL 常伴有肝脏浸润,临床可无任何表现,只是在做外周血常规检查和涂片镜检时偶然发现,具有独特的自然病程以及临床和生物学特征[2]。
【一般资料】
患儿女 16 天,因「其母孕 32 周发现腹腔占位」于 2017 年 2 月首次入院。患儿 G1P1,孕 38 周,剖宫,3280 g,羊水 III 度污染,Apgar 评分 7-9 分。孕 32 周时,B 超提示:腹腔占位,后逐渐增大。孕期产检羊水穿刺诊断「21 三体综合征」。出生发现腹胀,就诊于浙江平阳县人民医院,予以抗感染,监测生命体征等对症治疗,查腹部 B 超提示「右上腹囊实性混杂团块(畸胎瘤首先考虑)」,家长要求至温州医科大学附属第二医院就诊,查血常规示「WBC 61*109/L,N 16%,L 35%Hb 151 g/L,血小板 67*109/L」,原幼细胞 40%,查腹部 B 超提示「肝内高回声团 (血管瘤?):右侧腹腔混合回声团 (寄生胎可能)」,予以头胞哌酮舒巴坦联合青霉素抗感染治疗,建议进一步完善染色体、骨髓等检查,家长要求转来我院就诊,门诊查腹部 B 超提示「右侧腹腔非均质肿块 (96*80*56 mm):来自于腹腔后可能性大 (畸胎瘤声像?寄生胎不排外)」,为进一步诊治,于 2017 年 2 月至 2017 年 3 月在我院新生儿科住院治疗。入院查体:神清反应可,两眼距宽,外侧上斜,鼻根低平,颈短,双肺无异常,心前区可及Ⅱ级 SM 杂音,腹膨隆,右上腹可及直径约 10 cm*8 cm 肿块,根据影像学检查提示腹部巨大肿瘤,心脏超声存在房间隔缺损(Ⅱ)和动脉导管未闭,并且骨髓涂片及外周血免疫分型明确诊断为急性髓系白血病(图 1A、图 2A),既往羊水染色体检查诊断 21-三体综合征,诊断为「新生儿白血病?腹部畸胎瘤,21-三体综合征,先天性心脏病」。予置远红外辐射台、心电血氧监护,美平抗感染,碳酸氢钠碱化尿液等治疗;告知家长患儿病情复杂,住院期间腹腔肿瘤逐渐增大,外周血异常细胞比例逐渐增多、血小板及血色素逐渐下降,化疗并发症严重且预后极差,家长经考虑,要求放弃治疗并自动出院。
2017 年 12 月因「发现腹腔占位伴血象异常 10 月」该患儿第二次入院,第一次出院后在家中观察未予特殊诊治,患儿一般情况尚可,腹部包块亦无进行性增大,此次入院家长想做腹腔肿瘤的切除,门诊拟「腹膜后肿瘤(畸胎瘤可能)、急性髓系白血病」收住。入院后腹部 B 超仍提示腹膜后恶性肿瘤(畸胎瘤可能):患儿肿瘤巨大,甲胎蛋白不高,考虑成熟肿瘤的可能性偏大。复查骨穿涂片发现(图 1B)原幼粒 8.5%,早幼粒 2.5%,免疫分型中幼稚细胞表型虽为巨核细胞表型,但计数明显减少,仅 4.3%(图 2B ),竟达不到白血病的诊断标准。通过查阅文献,再综合该患儿合并新生儿唐氏综合征,符合唐氏综合征相关性的一过性骨髓增生异常髓系白血病的诊断;接着我们通过 NGS 检测 96 种 AML/MDS 相关基因突变,发现该患儿存在两种基因突变(图 3):TTN 基因:p.M1417L 位点突变以及 EPPK1 基因:p.A1408 G 位点突变。
【MICM 结果】
1、骨髓细胞形态学:第一次和第二次入院分别见图 1A 和图 1B
2017-03 (图 1A)原始细胞占 43%,该类细胞大小较一致,呈圆或毛刺状,胞核圆或类圆形,部分可见出芽现象。核染色质呈粗颗粒状,核仁可见或隐匿,胞浆灰蓝色不透明,周围有伪足样突起。 POX 染色:(-)100%; PAS 染色:(-)89%,(+)11% 阳性呈粗颗粒状。
2017-12 (图 1B) 原幼粒细胞约占 8.5%,早幼粒细胞约占 2.5%,巨系增生活跃,血小板散在可见。


图 1. 骨髓细胞形态学(A: 2017-03 B: 2017-12)
2、细胞免疫学:第一次和第二次入院流式分别见图 2A 和图 2B
2017-03(图 2A,外周血)
流式细胞仪计数 2 万个细胞,CD45dimSSC 稍高的异常细胞占比 34.4%,免疫表型:CD19-CD22-cCD79a-cCD22-cCD3-CD7+HLA-DR+MPO-CD33+CD13-CD11b+CD36+CD14-CD15-CD71+CD61-CD34+CD123-CD117+CD38+CD64-,考虑为髓系幼稚细胞。
A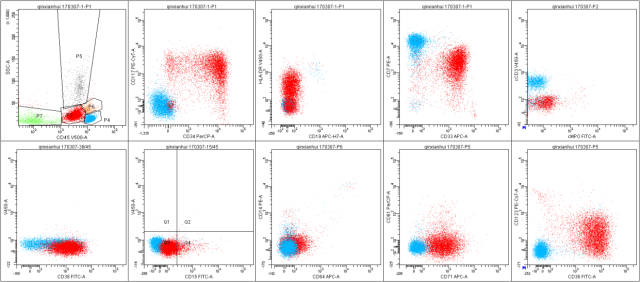
2017-12(图 2B)
流式细胞仪计数 2 万个细胞,CD45dimSSC 稍高,异常细胞占比 4.3%,免疫表型:CD19-CD22-cCD79a-cCD22-cCD3-CD7+HLA-DR+MPO-CD33+CD13-CD11b+CD36+CD14-CD15-CD71+CD61+CD41+CD34+CD123+CD117+CD64-,该群 4.3% 异常细胞考虑为急性巨核细胞表型。
B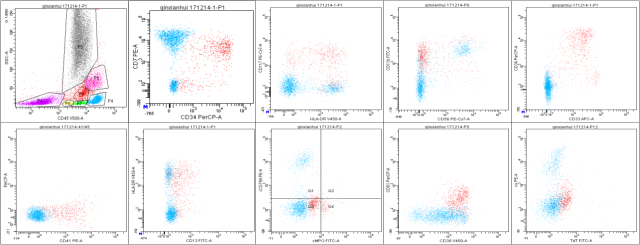
图 2. 流式细胞图(A: 2017-03 B: 2017-12)
3、细胞遗传学:
外院孕 32 周羊水穿刺:21 三体综合征。
4、分子生物学:
2017-12 第二次入院行 96 种 AML/MDS 相关基因检测(NGS 法)发现该患儿存在两种基因突变(图 3):TTN 基因:p.M1417L 位点突变以及 EPPK1 基因:p.A1408 G 位点突变。
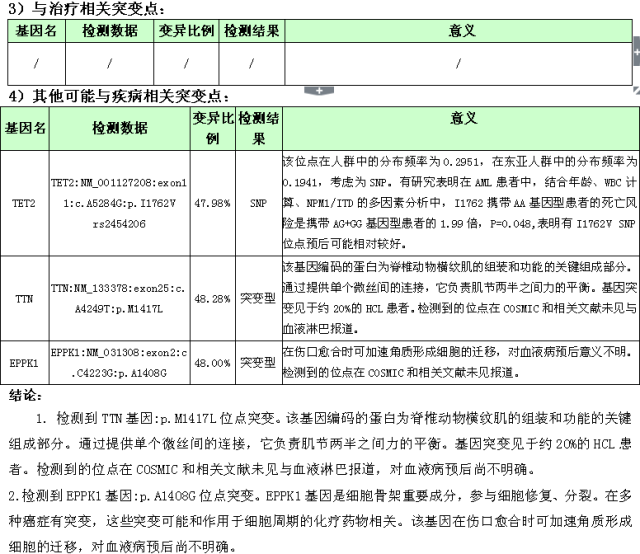
图 3 96 种 AML/MDS 相关基因检测(NGS 法)
【诊断】
唐氏综合征相关性的一过性骨髓增生异常髓系白血病
【治疗】
第二次入院后白血病治疗方面因无化疗指征,外科评估后于 2017 年 12 月行腹膜后巨大肿瘤切除术,术后病理提示:镜下:见肠组织、鳞状上皮、软骨、成熟脑组织、脉络丛、脂肪及纤维等组织,病理诊断为成熟性畸胎瘤。因此该患儿最后诊断为成熟畸胎瘤,一过性骨髓增生异常髓系白血病,唐氏综合征,房间隔缺损(Ⅱ)和 主动脉导管未闭,术后出院后至今失访。
【点评】
在唐氏综合征新生儿群体中,有一种特殊的血液学现象,即患儿出生时骨髓、外周血中出现大量幼稚巨核细胞,临床与急性巨核细胞白血病十分相似,不同的是这种幼稚巨核细胞可以自发消失。称为唐氏综合征相关性的一过性骨髓增生异常髓系白血病(Transient abnormal myelopoiesis TAM associated with down syndrome)[3]。此现象有两大特点:一是发生的时间较早,归属于新生儿白血病范畴;二是可以自发缓解。
本病例患儿第一次入院诊断急性髓系白血病、唐氏综合症、畸胎瘤等,因放弃治疗而出院,第二次是因为决定畸胎瘤的手术而意外发现骨髓和外周血中肿瘤细胞明显减少,且免疫表型中表达 CD41、CD61,符合巨核细胞的表型特点。该患儿诊断是历经了两次住院的演变才逐渐诊断明确的,正是因为患儿放弃治疗的巧合,畸胎瘤的存在,患儿得以第二次入院手术治疗,才「意外」发现包括形态、免疫分型等实验室检查的演变,并与血液科医师及时沟通以及查询文献,且结合病史患儿伴有唐氏综合征等最终锁定其诊断,并在 WHO2016 版血液肿瘤分类中找到了答案,隶属于急性髓系白血病及相关肿瘤分类中。
在对唐氏综合征相关性的一过性骨髓增生异常髓系白血病的研究中,人们发现编码红系/巨核系转录因子 GATAl 的基因突变广泛存在[4],GATA 1 基因位于 X 染色体上,编码锌指转录因子,对正常的红细胞和巨核细胞分化起着至关重要的作用[5],但该病例中患儿通过 NGS 检测 96 种 AML/MDS 相关基因,未发现,GATA 1 基因突变,而是存在 TTN 基因和 EPPK1 基因突变,这两种基因突变与唐氏综合征相关性的一过性骨髓增生异常髓系白血病的关系文献均未见报道。TTN 基因编码的蛋白为脊椎动物横纹肌的组装和功能的关键组成部分[6]。通过提供单个微丝间的连接,它负责肌节两半之间力的平衡。基因突变见于约 20% 的 HCL 患者。检测到的位点在 COSMIC 和相关文献未见与血液淋巴报道,对血液病预后尚不明确;EPPK1 基因是细胞骨架重要成分,参与细胞修复、分裂[7]。在多种癌症有突变,这些突变可能和作用于细胞周期的化疗药物相关。该基因在伤口愈合时可加速角质形成细胞的迁移,对血液病预后尚不明确,此两种基因与一过性白血病的关系,还有待于将来数据的积累。
尽管绝大多数该疾病可自发缓解,仍有高达 30% 的患儿会在未来的 1 至 4 年内发展成急性巨核细胞白血病。当然,极其罕见的严重类型可以直接导致死亡,最常见的死亡原因是由于肝脏大量原始巨核细胞浸润所引起的广泛肝脏纤维化、器官以及心肺功能衰竭[8]。
参考文献
[1]王东侠, 张文艺. 唐氏综合征与一过性白血病 [J]. 中国小儿血液与肿瘤杂志,2006,11(2):92-95.
[2]Roberts I, Izraeli S. Haematopoietic development and leukaemia in Down syndrome[J]. Br J Haematole, 2014, 22:2014.
[3]Rhoderick JA, Bradshaw WT.Transient myeloproliferative disorder in anewborn with Down syndrome[J]. Adv Neonatal Care, 2008, 8:208-218.
[4]Choi JK.Hematopoietic disorder in Down syndrome[J]. Int JClin Exp Pathol,2008 ,1(5):387–395.
[5]Reyes ZS, Bashir W, Pathare A. Transient Myeloproliferative Disorder and Down Syndrome: Is there a link[J].Sultan Qaboos Univ Med J. 2012 ,12(4):498-502.
[6]Chervinsky E, Khayat M, Soltsman S,et al. A homozygous TTN gene variant associated with lethal congenital contracture syndrome[J]. Am J Med Genet A, 2018 ,176(4):1001-1005.
[7]Ishikawa K, Furuhashi M, Sasaki T,et al. Intragenic copy number variation within human epiplakin 1 (EPPK1) generates variation of molecular size of epiplakin[J].J Dermatol Sci.,2018,1811(18):30234-2.
[8]Massey GV:, Zipursky A, Chang MN,et al. A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481[J]. Blood,2006 ,107(12):4606-4613.

夏敏,心血管医学硕士
儿内科副主任医师
从事儿科临床工作 20 余年,现任职于上海市儿童医院检验科,承担定期的儿内科专家门诊工作,参与本院血液肿瘤疑难病例的会诊,负责血液肿瘤流式和分子平台的工作,在儿童血液肿瘤方面,结合临床及形态、免疫、遗传和分子等予以综合和精准诊断。
曾作为主要承担者参与国家自然科学基金,海外青年合作基金课题,近年发表 SCI 和中华论著数十篇。