自从 2015 年美、中两国陆续站在国家战略层面的高度推出了「精准医疗计划」后,「精准医疗」四个字就一直受到了社会各界的广泛关注。
在上一期的稿件中,小编通过 OVA1® 这个成功的案例,讨论了 biomarker 开发的正式框架以及如何实现 biomarker 从实验室到临床的转化。而这一期的内容则主要包括在 biomarker 开发的 Phase I: Preclinical Exploratory Studies 中所需要用到的蛋白质组学技术。
Preclinical Exploratory Studies 的总体思路
在这个系列的第一讲中我们提到蛋白质生物标志物的发现策略主要有两种不同的思路。一种是和 GWAS 类似,通过大队列非靶向蛋白质组建设,去发现一些蛋白质表达量或是修饰状态的改变和疾病状态的相关性。而另一种是更为经典的生物标志物发现策略,即将整个过程分为不同的阶段,随着发现阶段的推进,不断增加样本数量并缩小目标蛋白,最终找到和疾病相关的一个或多个蛋白质生物标志物。
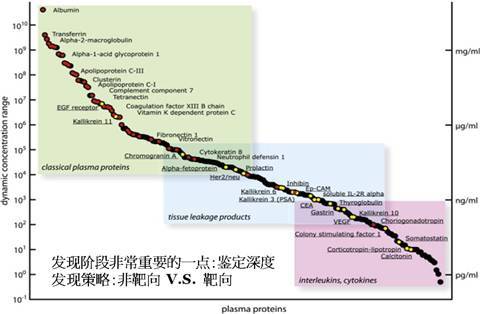
图 1. 血浆蛋白质浓度的动态范围
血液始终是我们去寻找疾病标志物的最佳样本之一,但从血浆中去发现 biomarker 却困难重重。尽管成人血浆蛋白质的浓度相对恒定(65-85 mg/mL),但血浆蛋白质的动态范围却非常大,通常横跨 10 到 12 个数量级(图 1)。而对疾病具有提示意义的标记物通常都是一些组织细胞的分泌蛋白,细胞破碎后的一些溢出蛋白等,这些蛋白质的含量通常都非常低。因此不管采用哪种发现策略,对血浆(或血清)样品进行合理的前处理以及质谱对血浆蛋白质组的鉴定深度都是成功发现 biomarker 的先决条件。
OVA1® 在发现阶段使用了 SELDI-MS 来寻找差异蛋白质,SELDI-MS 虽然分析通量很高,但由于受到蛋白芯片的制约,始终只能覆盖到血浆蛋白质的一小部分,这样我们在发现阶段就会错过太多有意义的信息,因此 OVA1® 的 panel 中都是一些丰度相对较高的蛋白质。
大队列非靶向蛋白质组建设依赖于强大的样本以及经费的资源,因此在这里我们主要讨论经典的生物标志物发现策略。经典的发现策略其实也存在着不同的思路,如果在最初的发现阶段我们没有任何的先验知识,那么我们就需要首先进行非靶向的蛋白质组学研究去发现疾病和正常的候选差异蛋白;但如果我们有来自文献或其他实验的先验知识,那么我们可直接采取靶向的蛋白质组学发现策略,通过不断的增加样本数量,缩小候选列表去得到最终的可能的 biomarker 蛋白。

图 2. 非靶向蛋白质组学生物标志物发现思路和技术方法
非靶向的蛋白质组学发现思路
非靶向蛋白质组学手段用于 biomarker 的发现已有十多年的历史,为了对该过程进行规范,有不少文献都总结了一个样本数递增,候选蛋白递减,从非靶向到靶向的一个发现策略[1]。总体来说,我们把临床前发现实验分为三个阶段: 差异发现,初步验证和最终确证(图 2)。根据我们上一讲中提到的生物标志物开发正式框架,我们整个临床前发现实验的目标是发现可信度高的生物标志物,此时还不需要确定最终用于临床检测的方法和判断疾病的筛选阈值。
1. 差异发现
在差异发现阶段,比起通量,我们更在乎的是血浆蛋白质的覆盖深度,只有在发现阶段能够定量到那些可能成为标志物的低丰度蛋白,我们才有可能在后续的实验中去评价它。
大家都知道蛋白质组学定量的方法有很多种,包括像基于体内代谢标记的 SILAC, 以及基于体外化学标记的稳定同位素二甲基标记,isobaric 标记等都可以在此过程中使用。由于缺乏合适的,和血浆蛋白组成类似的细胞系作为内标,SILAC 方法在血浆(或血清)蛋白质生物标志物的发现中很少应用[2]。而 isobaric 标记则提供了一个更为现实的血浆蛋白质定量方法。
Steven A. Carr 实验室进行了一项心血管疾病生物标志物的发现实验[3]。通过对肥厚梗阻型心肌病人实施酒精消融术,研究人员选取基准点,术后 10 分钟,60 分钟, 240 分钟四个点抽取病人外周血进行蛋白质组学分析来发现早期心肌损伤的标志物。为了增加对血浆低丰度蛋白的覆盖,研究人员采用 IgY14/SuperMix 免疫去除柱对血浆样品进行了高、中丰度蛋白的去除,蛋白质酶切后,采用 isobaric 标记来定量(图 3)。
在这篇文章中,研究人员得到了到目前为止对血浆蛋白质组的最为深度的覆盖,即便采用 2 条(及以上)的肽段来对蛋白质定量,依然在每个病人的四个采血时间点中有大于 4800 个蛋白质能够被定量,这些低丰度蛋白质的浓度范围跨越了 9 个数量级。像已知的早期心肌损伤标志物心肌肌钙蛋白 I 和 T(cTnI, cTnT)都可以非常高可信的在所有病人的血浆样品中被定量,要知道,这两个蛋白质在血浆中的含量一般都在 pg/mL 级。有了如此的定量深度,那么我们才能保证我们在最初的发现阶段不至于漏过太多的有潜在价值的标志物候选蛋白。
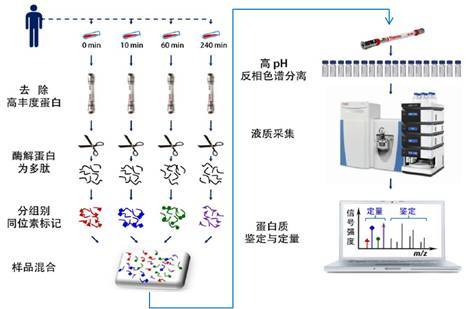
图 3. 早期心肌损伤标志物发现实验设计
其实我们在血浆中进行 biomarker 发现时会有一个假设,那就是我们认为肿瘤细胞的一些分泌蛋白,细胞表面蛋白以及溢出蛋白等会进入循环系统,从而成为疾病指征的标志物。而这些类型的蛋白质通常糖基化的程度都比较高,因此有不少的研究从糖蛋白入手,去发现血浆中的疾病标志物。
事实上,非常多的 FDA approve 的 biomarker 就是糖蛋白,例如前列腺癌标志物 PSA, 卵巢癌标志物 CA-125 等。再例如甲胎蛋白(AFP)有三种糖基化形式,AFP-L1,L2,L3。AFP-L3% assay 就是检测 AFP –L3 糖型在全部 AFP 中的比例,从而在长期肝病病人中指示肝癌的风险[4]。采用糖基化蛋白质组学手段进行血浆标志物的发现通常需要依赖各种凝集素(lectin)在蛋白质或者肽段水平对糖基化进行富集,随后采用定量蛋白质组学的策略去发现差异。
2014 年 MCP 上的一项研究就采用了这样的方法进行小细胞肺癌(SCLC)转移相关标志物的发现[5]。在这个工作中,研究人员分别采集了正常人群,非转移小细胞肺癌和转移小细胞肺癌病人的外周血用于标志物的发现。在实验阶段,病人的血浆通过除高峰度蛋白处理后,采用凝集素富集岩藻糖蛋白,在酶解后同样采用 isobaric 标记的方法进行糖蛋白的定量(图 4)。最后在 100 多个定量的糖蛋白中发现了潜在的小细胞肺癌的标志物。有些时候糖基化修饰的改变可能和蛋白质量的变化不一致。在此例中, PON1(Serum Paraoxonase 1)通过 western 验证蛋白质表达在肺癌病人中下调,而其糖基化却通过 hybrid lectin ELISA 发现在肺癌病人中上调。
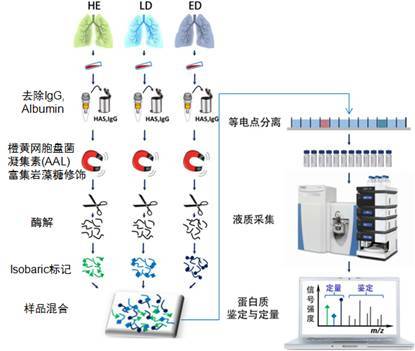
图 4. 小细胞肺癌糖基化标志物发现实验设计
2. 初步验证
通过发现阶段的实验,我们得到了一些差异候选蛋白,那么初步验证阶段的目的就是扩大样本数量对这些差异表达进行进一步的筛选。由于在差异发现阶段我们通常只会采用个位数的样本,有时还可能会采用混合样本,并且在差异发现阶段我们通常采用数据依赖的采集模式,那么这个时候很有可能某一个蛋白质在疾病和正常组中可能仅有 2,3 例样本有定量信息,这样我们无法采用统计学检验方法去判断这个蛋白是否是差异蛋白。而在初步验证阶段,我们一般会引入数十例样本,并且采用靶向或半靶向的方法进行蛋白质定量,以期尽量减少 missing value 的问题,使得每个靶蛋白都能在足够多例样本中定量,从而有足够的 statistical power 来判断差异蛋白。
在这个阶段我们可以采用多种靶向的定量方法,例如 inclusion list MS2(采用 MS1 定量), SRM, PRM 等靶向的方法,以及 DIA 等半靶向的方法。一般来说,我们的差异蛋白列表可能在 100-200 个蛋白质,那么这个时候我们需要考虑的就是质谱是否有足够的速度去覆盖这 100-200 个蛋白质的肽段。
一般来说,不管采用 inclusion list MS2, SRM 还是 PRM, 我们检测一个肽段通常需要 50-80 毫秒,那么一个 2 秒的 cycle 就能检测约 30 个肽段,若想要覆盖 200 个蛋白质的肽段,我们就需要一定长度的色谱洗脱时间,然后分段监控不同的靶肽段。而在样品前处理上,为了能对低丰度蛋白质定量,通常还是需要进行高丰度,乃至中丰度蛋白的去除,但是由于样本量增加,我们一般不再进行肽段的预分级。若有些蛋白丰度确实太低,即便再进行了高、中丰度蛋白质去除和靶向检测后仍然不能定量到该蛋白,且这个蛋白质又足够重要,那么我们还是需要去进行肽段预分级,但只需要对这个肽段所在的组分进行检测即可。
此外,在是否需要掺入重同位素肽段作为内标这一点上,由于差异蛋白数量较多,从经济上考虑一般不适合做重标掺入,毕竟肽段合成用的重同位素氨基酸价格不菲。我们可以考虑采用总离子流强度,或是一些 house keeping 蛋白来做样品间的归一化。
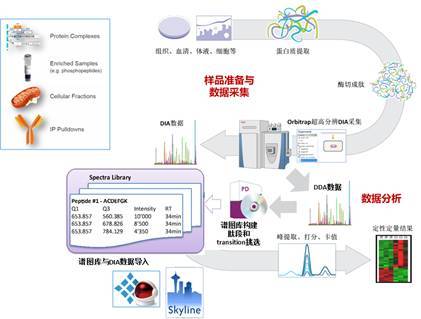
图 5. DIA 工作流程
DIA 和 SRM, PRM 不同,我们称之为半靶向的采集方式,其数据采集还是非靶向鸟枪法的模式,而在数据分析谱图库构建时我们却可以选择性的去构建该库。相比于 DDA,DIA 的优点在于可显著减小 missing value 的问题,使得每个定量的蛋白质几乎可以在所有样本中都定量。
根据谱图库构建方式的不同,DIA 的方法可以用于整个发现流程的不同阶段。若我们将 DDA 采集的所有鉴定蛋白都用于构建谱图库,那么这样的 DIA 方法适用于差异发现阶段。我们也可以只将发现阶段的候选差异蛋白用于构建谱图库,那么这种半靶向的 DIA 则可以用于初步验证阶段。
2015 年 JPR 的一篇文章对于尿液样本采用 DIA 的方法来寻找差异蛋白,通过高通量的自动化样品处理流程,一个样本只需 2.75 小时便能到实验结果,其中还包括 30 分钟的质谱分析时间(图 5)。结果在通过 DDA 数据建得的包含 2500+个蛋白质的谱图库中,有大于 1300 个蛋白能通过 DIA 数据在单个样品中定量[6]。由此我们可见,DIA 方法的优点是简单快速,为了达到这个目的通常在做 DIA 实验时我们不对样品进行肽段预分级。当然在血浆样品中为了进一步覆盖更多的低丰度蛋白,必要的高、中丰度蛋白去除也是不可避免的,我们在 demo 实验室也进行了这样的 DIA 血浆蛋白质组实验,得到了不错的效果,具体请咨询周岳工程师(yue.zhou@thermofisher.com)。但对于血浆蛋白质来说, single shot 的 DIA 实验始终还是存在着覆盖深度不够的问题,因此在进行 DIA 实验时,肽段的预分级有时也是很有价值的。
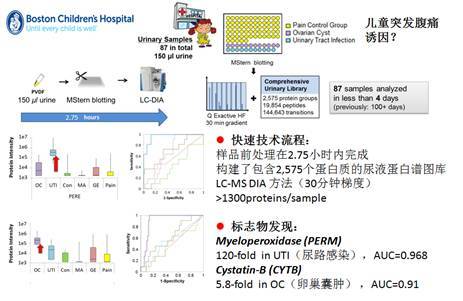
图 6. 尿液样本 DIA 分析用于发现差异蛋白
3. 最终确认
通过上一步的筛选,这时我们的差异蛋白列表会进一步缩小,一般包含十个到几十个的候选蛋白。那么在最终确认阶段,我们则要通过百例以上的样本来进一步在大样本上验证差异蛋白,并对最终确认的这些差异蛋白质构建预测模型,评价这些 marker 对于疾病判断的真阳性率,假阳性率,ROC 曲线等指标。
在这个步骤中我们可以采用基于质谱的靶向定量方法,或直接使用抗体方法(如 ELISA)来对差异蛋白进行定量。采用抗体方法定量的优点是灵敏度高,但是由于抗体的质量往往无法保证,因此很有可能造成假阳性或假阴性的结果。质谱靶向定量方法 SRM,PRM(尤其是 PRM)的方法具有非常高的选择性,但是灵敏度却不如抗体方法, 对于血浆中丰度很低的蛋白质(ng/mL 级)往往无法检测。对于这种情况,我们通常会在蛋白质或者肽段水平对我们的目标蛋白先进行免疫富集,随后再进行 SRM,PRM 的分析,这样可以同时满足高灵敏度和高选择性。而在最终确认这一步中,在进行 SRM,PRM 实验时,我们一般需要掺入重标肽段作为内标来对目标蛋白进行绝对定量。这样的 immuno-SRM, immune-PRM 的方法其实有许多商业化的解决方案,包括经典的 SISCAPA 方法[7],以及 Thermo Fisher 的 MSIA 方法[8] 等。
PRM 方法可能大家不一定非常熟悉。PRM 称为平行反应监控(Parallel Reaction Monitoring),其是在 Q-Orbi(QE, Fusion 系列)上实现的一种靶向定量采集方法。在 PRM 模式下,我们对 inclusion list 中的目标母离子进行捕获和碎裂,用 Orbitrap 采集包含所有碎片离子的二级谱,而在数据分析时则和 SRM 类似,采用特定的母子离子对(transition)来对目标肽段定量。因此,PRM 和 SRM 最大的差别在于,SRM 的母离子和碎片离子都是低分辨的,而 PRM 的母离子是低分辨的,而碎片离子是高分辨的(图 7)。这样,PRM 就能比 SRM 更能不受基质效应的干扰,经数据实测,PRM 能在灵敏度上和 SRM 相当,而在选择性上优于 SRM。其实,PRM 最大的优点还是在于经济性上,当实验室只有一台高分辨质谱时,我们也能完成标志物开发 Preclinical Exploratory Studies 的上下游全部流程。
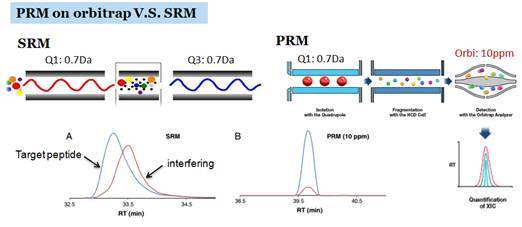
图 7. SRM, PRM 方法示意
当通过上述三个步骤,我们最后得到了高可信的 biomarker candidate, 并且通过预测模型对这些 marker 的 clinical performance 进行了初步评估后,我们就能够很有信心的进入 Phase II study, 真正去建立最终用于临床检测的方法。在 Phase II 的研究中,不管是对抗体还是质谱方法,我们对其 analytical performance 都会有更严苛的要求来真正达到临床检测的需求(具体请参考第二期)。
下期预告
在本期中,小编主要和大家聊了一下标志物发现 Phase I: Preclinical Exploratory Studies 中的非靶向的蛋白质组学发现思路。那么下期中我们将会继续关注 Phase I study, 去进一步了解一下另一种发现思路:即靶向的蛋白质组学发现策略。
特此鸣谢撰稿人——唐家澍工程师
参考文献
1. Rifai,N., M.A. Gillette, and S.A. Carr, Proteinbiomarker discovery and validation: the long and uncertain path to clinicalutility. Nat Biotechnol, 2006. 24(8):p. 971-83.
2. Mangrum,J.B., et al., Intact stable isotopelabeled plasma proteins from the SILAC-labeled HepG2 secretome. Proteomics,2015. 15(18): p. 3104-15.
3. Keshishian,H., et al., Multiplexed, QuantitativeWorkflow for Sensitive Biomarker Discovery in Plasma Yields Novel Candidatesfor Early Myocardial Injury. Mol Cell Proteomics, 2015. 14(9): p. 2375-93.
4. Li,D., T. Mallory, and S. Satomura, AFP-L3:a new generation of tumor marker for hepatocellular carcinoma. Clin ChimActa, 2001. 313(1-2): p. 15-9.
5. Ahn,J.M., et al., Integrated glycoproteomicsdemonstrates fucosylated serum paraoxonase 1 alterations in small cell lungcancer. Mol Cell Proteomics, 2014. 13(1):p. 30-48.
6. Muntel,J., et al., Advancing Urinary ProteinBiomarker Discovery by Data-Independent Acquisition on a Quadrupole-OrbitrapMass Spectrometer. J Proteome Res, 2015. 14(11): p. 4752-62.
7. Anderson,N.L., et al., Mass spectrometricquantitation of peptides and proteins using Stable Isotope Standards andCapture by Anti-Peptide Antibodies (SISCAPA). J Proteome Res, 2004. 3(2): p. 235-44.
8. Krastins,B., et al., Rapid development ofsensitive, high-throughput, quantitative and highly selective massspectrometric targeted immunoassays for clinically important proteins in humanplasma and serum. Clin Biochem, 2013. 46(6):p. 399-410.