自从 2015 年美、中两国陆续在国家战略层面高度推出了「精准医疗计划」后,「精准医疗」四个字一直受到了社会各界的广泛关注 !
首先我们来明晰一下精准医疗的定义,精准医疗是指根据患者的临床信息,应用现代遗传技术、分子影像技术、生物信息技术,结合患者的生活环境和方式,实现精准的疾病分类及诊断,制定具有个性化的疾病预防和治疗方案。其中非常重要的一点就是建立遗传信息和疾病之间的联系,从而来对疾病进行诊断,指导治疗方案和判断病人的预后。随着基因测序技术的发展,基因水平的遗传信息和疾病的关联最早被人们所关注。
GWAS (Genome-Wide Association Study) [1], TCGA (The Cancer Genome Atlas)[2] 等项目通过大队列的相关性研究,致力于建立基因的表达量、拷贝数、点突变以及表观遗传学信息的改变和各种疾病,尤其是癌症之间的关系。但中心法则告诉我们,基因水平的信息告诉我们的是疾病发生的概率(即可能性),而蛋白质作为生命活动的最终执行者,其状态的改变能更为直接的反应疾病的发生、发展状况。事实上,蛋白质作为生物标志物相比于基因来说,其应用历史更为久远。

图 1. PRECISION MEDICINE INITIATIVE
目前,就蛋白质生物标志物的发现策略来说,也呈现出多种不同的思路。一种是和 GWAS 类似,通过大队列非靶向蛋白质组建设,去发现一些蛋白质表达量或是修饰状态的改变和疾病状态的相关性。而另一种是更为经典的生物标志物发现策略,即将整个过程分为不同的阶段,随着发现阶段的推进,不断增加样本数量并缩小目标蛋白,最终找到和疾病相关的一个或多个蛋白质生物标志物。基于质谱的蛋白质组学一直被认为是生物标志物发现的重要手段,但截止到目前为止,仅有一个标志物是通过蛋白质组学手段发现的。
在这系列的文章中,小编将和大家深度探讨一下如何逾越从标志物发现到临床转化的鸿沟。这四期的内容包括:
(1) Biomarker 的市场现状和发展趋势,FDA 对于 Biomarker 的申报流程
(2) 目前唯一一个通过蛋白质组学手段发现的 Biomarker – OVA1® 从发现到 FDA clearance 的转化之路
(3) Biomarker 发现各阶段的技术手段
(4) 采用 LDTs 的形式进行的临床质谱体外诊断的市场现状和应用实例
Biomarker 的市场现状和发展趋势
FDA 是以器械 (device) 的形式来对 biomarker 进行审批的,即不同的生产厂家可针对同一个 biomarker 推出自己的不同的检测方法。FDA 中的 CDRH (Center for Devices and Radiological Health) 负责受理这些检测器械的申报和审批。
小编之前对 FDA 已经批准的肿瘤生物标志物进行了汇总统计,主要包含三类的生物标志物:
(a) 蛋白质生物标志物:包含了针对 26 个 biomarker 的 63 个检测方法,例如大家所熟知的 PSA, CEA, HER-2 等,其中还含有一个多标志物检测方法(biomarker panel)- OVA1®。目前这些检测基本都是以免疫学检测的方法灶进行,用于检测的样本则包含了血浆、肿瘤原发灶、转移灶等多种类型的生物学样本。
(b) 基因水平的生物标志物:该水平的标志物包含的检测类型更为多样,例如检测 mRNA 的表达量、基因拷贝数、基因点突变以及表观遗传学的改变等。目前 FDA 已批准了针对 14 个 gene biomarker (一个 biomarker panel 算一个 marker) 的 23 个检测方法,其中包含四个 biomarker panels, 分别是: MAMMAPRINT, Cologuard, GeneSearch breast lymph node (BLN) assay, Prosigna。
(c) 细胞水平的生物标志物:目前 FDA 批准了一个 CTC 的检测方法,即 CellSearch Circulating Tumor Cell Kit。该检测用于评估已发生转移的前列腺癌、乳腺癌和结肠癌病人的预后状况。
在 2003 年以后,FDA 每年批准上市的检测方法约有 50 个, 而新的 marker 每年仅有 3 个。这主要源自于新的 biomarker 的成熟周期非常漫长,平均而言,一个新的 Biomarker 从最初的实验室发现到最终被 FDA 批准上市需要花 15 年的时间。 因此,业界普遍认为,在人类基因组计划完成后的这一个十年才会是新的 biomarker 的集中爆发期。
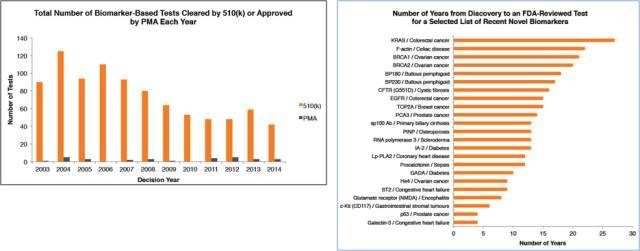
图 2. Biomarker-Based IVD Tests 市场趋势摘要
FDA 对于 Biomarker 的申报流程
FDA 根据检测风险的高低将医疗器械(medical device)分为三类,这里的检测风险是根据检测方法的用途和错误结果对病人可能造成的危害来做判断。
第三类 device 是风险最大的一类,该类 device 需要通过 FDA 的上市前许可(premarket approval, PMA)途径来完成审批上市。在 PMA 的标准中,供应商必须通过荟萃分析或临床研究证明该 device 对病人的疾病判断是有效且有临床获益的。
第二类 device 是中等风险的 device, 由 FDA 通过 上市前通知(premarket notification)的方式来进行审批, 即我们常说的 510(k)。510(k) 是一个等效性申明,即供应商只需证明新的 device 相比于现有 device 的等效性即可。根据被分析物的不同,510(k) 的申请不一定需要临床研究。但是还有一类特殊的情况,即对于中等风险的 device, 但没有现有的 device 可比对,CDRH 允许供应商通过一个特殊的 510(k) clearance, 常被称为「De novo」pathway 来进行申报。该途径需要证明 device 的安全性和有效性, 但不需要经历第三类 device 的 premarket inspection, 相比 PMA 会更快。大多数的新的 biomarker 都以这种方式被 FDA 批准上市。
在大致了解了 biomarker 申报的类别之后,小编再和大家简单过一下 FDA 对于体外诊断设备的审批流程。
第一步:供应商准备 pre-submission application 提交给 FDA,也就是大家所知道的 pre-IDE。通过这一步呢,供应商通知 FDA 我有一个新的医疗检测设备需要上市,而在这份文件中需要包含该检测方法的预期用途,技术方法的描述, 分析实验和临床实验的计划。
第二步:通过和 FDA 的磋商,供应商和 FDA 对新的检测设备的用途和实验计划达成一致,并确定是通过 PMA, 510(k) 还是 de novo 的途径上市。
第三步:供应商完成必要的分析实验和临床实验。其中高风险的临床试验需要事先通过 Investigational Device Exemption (IDE) application 报备 FDA。
第四步:供应商向 FDA 提交 510(k) or PMA 申请。CDRH 在指定时间内完成审核。510(k) 的审核时间为 90 天, PMA 为 180 天。
第五步:若 FDA 发现申请材料的不足,则会向供应商提出意见,申请搁置,供应商需要补充实验或其他一些资料直至达到 FDA 的要求。
第六步:最终的上市前准备。对于第二类 device, 在 FDA clear 后可以在美国上市,并根据检测的复杂程度决定在哪种 CLIA-certified 实验室中进行检测。若要在家,诊室或其他 non-CLIA 实验中进行这些检测, 则还需要提供其他一些资料。对于第三类 device, FDA 根据供应商提供的数据决定 device 的安全性和有效性。此外,FDA 另外还需要对公司进行 premarket inspection, 以决定该公司的生产条件达到了 21 CFR 820 的质量标准。以 PMA 形式上市的 device 有着更为严格的上市后责任,任何生产或设计上的改变都需要每年向 FDA 报备,任何可能影响安全性和效果的生产或设计上的改变都需要向 CDRH 提交 PMA 补充申请。
下期预告
在本期中,小编带着大家简单浏览了一下 Biomarker 的市场现状和发展趋势以及 FDA 对于 Biomarker 的申报流程。那么下期中我们将会探讨一下针对 FDA 的申报要求我们如何组织我们的 biomarker 发现实验,此外我们还会通过 OVA1® 的成功案例来具体,详细的分析一下蛋白质组学用于 biomarker 发现的成功经验。
特此鸣谢撰稿人---唐家澍工程师
参考文献
1. Manolio, T.A., Genomewide association studies and assessment of the risk of disease. N Engl J Med, 2010. 363(2): p. 166-76.
2. NIH Launches Cancer Genome Project Washington Post Dec 14, 2005