Orbitrap 自发明起,就一直是科学家实现世界顶级科研突破的有力伙伴。
今天就让我们来一探全球顶尖 PI 最近发表的文章和技术成果,看看 Orbitrap 技术是如何助力顶级科研的:
Matthias Mann
基于 Orbitrap 的全新方法学研究,创新开发 BoxCar 数据采集方式
为了应对蛋白质组学中的动态范围挑战,Mann Lab 最近开发了 「BoxCar」的数据采集方法(Meier et al., Nat. Methods, 2018),这显著提高动态范围大的样本中的蛋白鉴定深度,例如血浆或组织样本(Geyer et al., Cell Syst., 2018, Doll et al., Nat Commun., 2017)。
Orbitrap 质谱仪在灵敏度和采集速度方面取得了很大进展,使蛋白质组覆盖深度范围越来越广。然而,这些进展主要局限于 MS²水平,而用于 MS¹扫描的离子采集仍然非常低效。Mann Lab 介绍了一种数据采集方法,称为 boxcar,一级全扫时采用分段累积的方法,使得平均离子注入时间相较标准全扫描增加 10 倍以上。对一个人类癌细胞系进行 1 h 分析,该方法鉴定到之前在 24 个组分中鉴定到的 90% 以上的蛋白质,并且定量到了 6200 多个蛋白。在小鼠脑组织中,仅在 100 min 内就检测到超过 10000 种蛋白质,并将灵敏度扩展到低阿摩尔级。

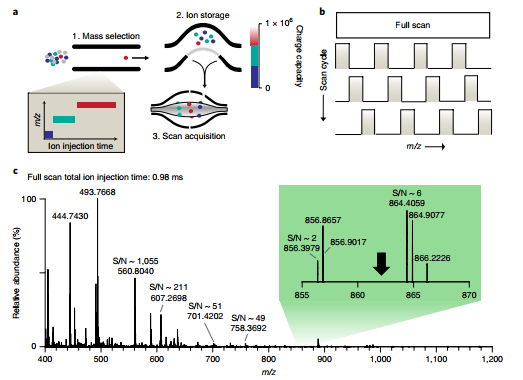
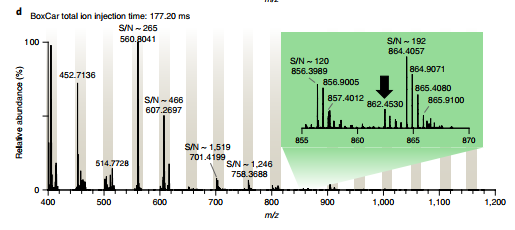
如今 Boxcar 技术已经全面搭载于全新的:

Thermo Scientific™ Orbitrap Eclipse™
三合一质谱平台

Thermo Scientific™ Orbitrap Exploris™ 480
组合型高分辨率质谱仪
Matthias Mann
多组学研究进展:为建立调控潜能性状态转变的模型机制奠定了基础

多潜能干细胞是高度动态且持续进展的,多潜能性的 naïve 和 primed 两种状态之前已经有深入报道,但是对于两种状态之间的转换过程的研究,却仍然是不完整的。文章剖析了胚胎从着床前到着床后胚层分化的多能态转变动力学,通过对蛋白质组、磷酸化蛋白质组、转录组和基因组的综合分析,发现磷酸化蛋白质组的快速、急性和广泛变化等特点,先于基因组、转录组和蛋白质组的有序变化。文章奠定了调控潜能性状态转变模型的基础,对多潜能性的多层控制提出了全新见解。
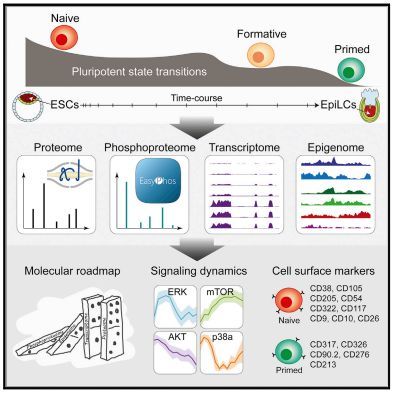
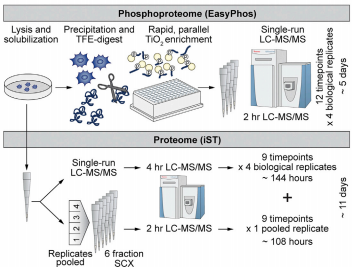
Matthias Mann
蛋白质组学助力卵巢癌标志物新靶点发现

该篇发表在 Nature 上的文章介绍了一种全新技术:通过将激光捕获显微切割技术与基于 Orbitrap 的高灵敏蛋白质组分析技术相结合,从 11 位高级别浆液性卵巢癌 (HGSC) 病人石蜡包埋组织中提取了 107 个癌症与基质细胞,随后进行蛋白质组分析, 指出与肿瘤转移密切相关的成纤维细胞 (cancer-associated fibroblast,CAF) 中调控蛋白 N-甲基转移酶 (N-methyltransferase(NNMT)) 是卵巢癌发生、发展以及转移的关键调控因子,可能成为全新治疗靶点,未来同样可能为造福 HGSC 病人的福音。
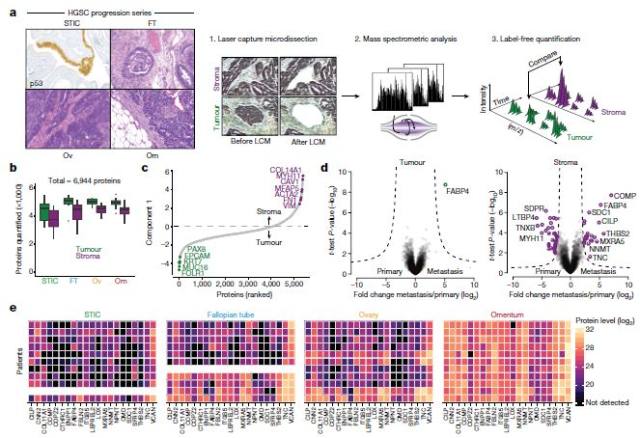
此研究基于 Q Exactive 和 Q Exactive HF
Oliver Fiehn
非靶向代谢组学中质谱结构注释有所突破

尿液代谢物经常被用于许多临床和生物医学研究,但通常仅限于少数经典化合物。其实,代谢组学分析可以检测到更多的代谢信号,可以用来精确定义个人的健康状况。然而,许多化合物仍然未被鉴定,妨碍了得出相关生物化学结论。
在这篇文章中,Fiehn Lab 用基于 HILIC-Q Exactive HF 质谱和 C18-Q Exactive HF 两种非靶向代谢组学分析方法,检测到的所有代谢物。检测到 9000 多种代谢物,其中 42% 的化合物有 MS/MS 信息。采用标准品经过精确质量数、保留时间和二级信息鉴定了 175 种化合物。用一级和二级信息,鉴定到另外 578 个化合物。
Steven Gygi
FAIMS 方法的多重定量表征与优化

在定量蛋白质组学中,同位素标记法是提高蛋白组定量通量、精确度的有力技术。然而,定量的动态范围和准确度可能会因标记肽段共隔离的限制,致使肽段释放的报告离子被合并定量。通常采用在线或离线过滤的方式来减轻共隔离的干扰,但是往往会导致蛋白质和肽段鉴定的缺失。为了解决这一问题,本文提出了一种高场非对称波形离子迁移质谱 (FAIMS) 方法,可以减少前体离子共流出、提高多重定量准确度和动态范围。在不牺牲蛋白质鉴定数量的前提下,FAIMS 有力地提高了基于高分辨率 MS²(HRMS²)和 SPS-MS³的定量准确度。经过进一步优化条件,使 FAIMS 更加稳健并提供参考方法,推动 FAIMS 进一步提升同位素标记定量的能力。
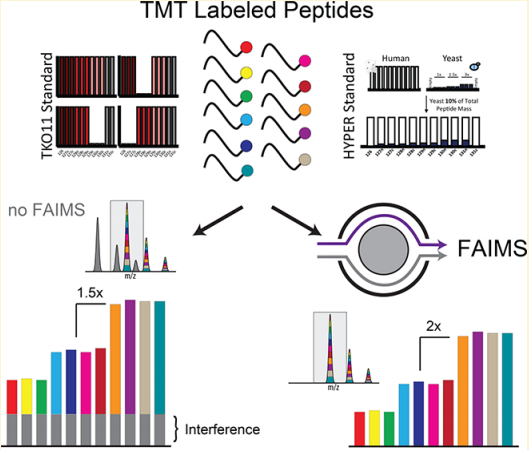

全新的 Thermo Scientific™FAIMS Pro™ 接口
Bing Zhang
结合蛋白基因组与修饰蛋白组学研究,全面剖析结肠癌
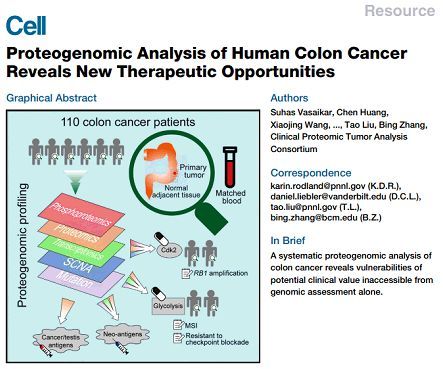
蛋白基因组学 (Proteogenomics) 是利用蛋白质组学数据,尤其是高精度的串联质谱数据, 结合基因组和转录组数据对基因组进行注释。除此之外,蛋白质组数据还能系统发现蛋白质特有的翻译后修饰、可变剪接等信息。Orbitrap 质谱兼具高精度、高灵敏度和高稳定性等优势,可为研究人员提供强有力的生物质谱技术,已然成为蛋白基因组学研究不可或缺的一部分。
本项研究中,研究人员收集了 110 例结肠癌样本。研究通过对来自 110 个结肠癌病人的肿瘤样本、临近正常组织(NATs)和血液样本,进行蛋白质组学、全外显子测序、RNA-seq、miRNA-seq 研究。为了进一步探究肿瘤和正常组织中的蛋白质组差异,作者还结合 TMT 标记定量和磷酸化蛋白质组分析,总结肿瘤的临床和病理特征。研究证实,这些基因变异确实伴随着蛋白质组/磷酸化蛋白质组学的变化。研究人员利用基因组、蛋白组和修饰组学相结合的分析策略,首次对结肠癌的蛋白基因组进行了全面的剖析,为结肠癌研究提供了新的研究思路。
随着组学研究的不断深入,质谱技术,尤其是高分辨质谱技术能够助力顶级 PI 在方法学研究、技术突破、精准医疗等各个领域取得新进展。Orbitrap 自发明以来,有越来越多不断探索技术极限的科学家,选择 Orbitrap 成为研究之路上的伙伴;随着时间的推移,正是越来越多科学家的认可,造就了 Orbitrap 如今组学研究领域金标准的地位,也成为顶尖 PI 创新研究的共同选择。希望下个 20 年,Orbitrap 能为科学家们带来更具创新性、更突破极限的助力,一起携手,让世界更健康、更清洁、更安全。
本文内容来自各 PI Lab 自有网站及 PI 近期文献整理
参考文献
[1] BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nature Methods. 2018.
[2] Multi-omic Profiling Reveals Dynamics of the Phased Progression of Pluripotency. Cell Systems. 2019.
[3] Structure Annotation of All Mass Spectra in Untargeted Metabolomics. Cell Systems. 2019.
[4] Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature. 2019.
[5] Structure Annotation of All Mass Spectra in Untargeted Metabolomics. Anal Chem. 2019.
[6]. Vasaikar S, Huang C, Wang X, et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell. 2019