2018 年,O 药、K 药相续上市以及诺贝尔医学奖的颁布,使得癌症免疫检查点阻断治疗成为医学及科研圈的热门话题,媒体、研究机构以及医疗和制药企业通过网站、微信、微博等途径报道了大量与其研究进展相关的文章,至此,免疫检查点阻断治疗作为癌症治疗的可靠方法,走进了大众的视野,为少数癌种的治愈带来希望。
然而,患者响应率低、容易产生治疗抵抗(即耐药)是目前该疗法遇到的瓶颈。2018 年 12 月 17 日,《Nature》刊登了一篇克服免疫检查点阻断治疗抵抗的新策略。小编将从免疫检查点阻断疗法的优势与局限性、新策略的提出、ADAR1 缺失对肿瘤的影响及机制三方面为大家深度剖析这一新策略。

一、免疫检查点阻断疗法虽然靠谱,但不是万能的
近年来,癌症免疫治疗中的免疫检查点阻断治疗凭借着阻断免疫检查点,激活内源性抗肿瘤 T 细胞(开启机体天然屏障),杀死癌细胞的优势,在众多的抗癌疗法中脱颖而出,取得了许多可喜的成绩,可谓是「抗癌明星」。
然而,这一治疗方法并不是万能的。在临床医生和科学家积累了大量应用免疫检查点阻断疗法进行癌症治疗的成功经验的同时,也发现了该疗法的局限性,即:大部分患者对免疫检查点阻断治疗无响应或产生治疗抵抗。
二、研究者将目光投向 ADAR1
为了克服癌症患者对免疫检查点阻断治疗的抵抗,来自美国丹娜法伯癌症研究院儿科肿瘤学系的 Jeffrey J. ishizuka 等研究者利用成簇的规律间隔的短回文重复序列(CRISPR)基因组编辑技术,鉴定 B16 黑色素瘤移植模型的基因表达,确定当某个基因敲除时,活体动物对免疫治疗的敏感性。通过该方法鉴定出多个对内源性 RNA 具有潜在修饰作用的基因,其中核糖核酸腺苷脱氨酶 1(ADAR1)引起了研究者关注。
既往研究表明,ADAR1 能够结合并作用于内源性双链 RNA(dsRNA),抑制 dsRNA 应激。此外,ADAR1 介导的腺苷到肌苷(A-to-I)的 RNA 编辑是抑制干扰素(IFN)反应的一个关键步骤,而肿瘤干扰素及其相关通路的激活则是影响肿瘤免疫治疗的一个重要因素。基于此,研究者设计了系列试验,以探索 ADAR1 缺失对肿瘤的影响及机制。
三、ADAR1 缺失对肿瘤的影响及机制
1. 使多个移植性肿瘤动物模型对免疫检查点阻断疗法高度敏感
研究者通过 CRISPR 基因编辑技术制备了 ADAR1 p150 亚型或 ADAR1 p110/p150 亚型缺失(以下简称 Adar1-null)的 B16 小鼠黑色素瘤模型。研究发现,Adar1-null 野生型 B16 肿瘤以及其他 3 个移植性肿瘤模型的 Adar1-null 肿瘤细胞系对程序性细胞死亡蛋白-1(PD-1)免疫疗法高度敏感。
2. 导致肿瘤免疫微环境的重塑
研究者比较了 Adar1-null 肿瘤和未接受治疗的野生型 B16 肿瘤的免疫微环境,结果发现,Adar1-null 组的 CD3+T 细胞、CD4+T 细胞、CD8+T 细胞、γδ T 细胞、自然杀伤细胞(NK)的比例显著增加;骨髓来源的抑制性细胞(MDSC)和肿瘤相关中性粒细胞显著减少。免疫细胞表达的趋化因子平衡发生了变化。IFNβ和 IFNγ蛋白水平升高。
3. 在 IFN 刺激下,引起肿瘤细胞生长停滞和凋亡
研究者通过 IFN 刺激试验发现,IFNβ或 IFNγ刺激使 Adar1-null 细胞存活率明显降低,凋亡率增加。在 CT26 和 Braf/Pten 肿瘤中也观察到类似的结果。当采用 IFNβ或 IFNγ培养时,Adar1-null 细胞中的细胞因子和趋化因子基因(如 Ifnb1、Il6、Ccl5、Cxcl9 和 Cxcl10)表达上调。
4.Adar1-null 肿瘤对免疫治疗的敏感性需要 IFN 的介导
研究者对 Adar1-null 肿瘤细胞系进行了 Ifnar2、Ifngr1 或 Stat1 的双敲除,以及同时敲除 Ifnar2 和 Ifngr1 的三敲除,发现:经 IFNβ和 IFNγ刺激的 Adar1-null 肿瘤,当同时敲除 Ifnar2 和 Ifngr1 时,消除了体外生长停滞和 IFNβ分泌表型。敲除 Stat1 也能得到类似的结果。提示:Adar1-null 肿瘤对免疫治疗的敏感性需要 IFN 的介导。
5. 使肿瘤对放射治疗敏感
体外试验表明,接受辐射治疗后,Adar1-null 组 TNFβ分泌量显著升高。进一步研究发现,Adar1-null 组动物在接受剂量为 12.5Cy 的放射治疗或应用咪喹莫特的传统治疗后,体内肿瘤的生长抑制明显,生存率显著提高。
6.ADAR1 编辑的 RNA 优先被 IFN 诱导
研究者发现,在 IFNβ刺激下,ADAR1 介导 A-to-I 编辑的 RNA 显著上调,临近的编辑位点与可及染色质 IFN 诱导区高度相关。提示:IFN 作为 dsRNA 传感器的配体,增加了 ADAR1 介导的 RNA 的转录。
7.PKR 和 MDA5 介导不同的表型
研究者采用 CRISPR 技术进行的全基因组编辑,发现,蛋白激酶 R (PKR)或黑色素瘤分化相关基因 5(MDA5)介导的 dsRNA 应激可以增强 Adar1-null 肿瘤对免疫检查点阻断的响应。其中,PKR 介导的 dsRNA 应激是 IFN 诱导的 Adar1-null 肿瘤细胞生长停滞的主要机制;MDA5 介导的 dsRNA 应激则是增加 Adar1-null 肿瘤炎症所必需的。
8. 诱导足够的炎症, 绕过 CD8+T 细胞识别癌细胞的免疫治疗机制
因免疫检查点阻断治疗起效,需要 CD8+T 识别癌细胞。研究者首先制备了免疫检查点阻断治疗抵抗模型(即:CD8+T 细胞不识别肿瘤的模型),通过比较 Adar1 和 B2m 缺失的细胞系与仅 B2m 缺失的细胞系对粒细胞-巨噬细胞集落刺激因子(GM-CSF)分泌的全肿瘤细胞疫苗(GVAX)和 PD-1 阻断的体内免疫治疗的敏感性,发现:ADAR1 的缺失,使得免疫治疗消除了大量的 B2m-null 耐药肿瘤,恢复了免疫治疗的敏感性。其他几个获得性免疫治疗抵抗模型(敲除 H2-K1、Nlrc5 和 Jak2 等)的试验也得出类似的结果。提示:在 CD8+T 细胞不识别肿瘤细胞的情况下,Adar1 缺失同样能克服免疫治疗抵抗。
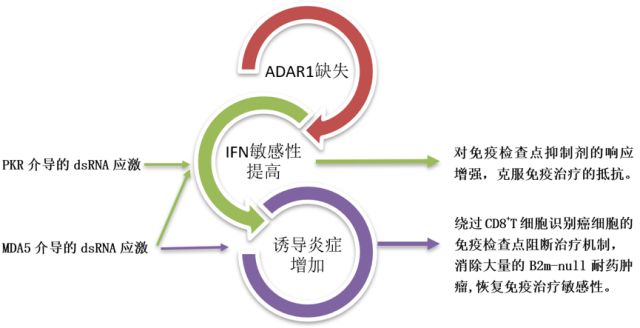
图 ADAR1 克服免疫检查点阻断治疗抵抗的主要机制
目前,免疫检查点阻断疗法在临床上取得了令人振奋的肿瘤治疗效果,成为当今最受瞩目的肿瘤免疫疗法之一。然而,免疫检查点抑制剂仅对部分肿瘤患者和部分类型的肿瘤有效,其治疗的有效率和癌种有待进一步提高和扩大。
传统的免疫检查点阻断疗法,需要细胞毒性 CD8+T 细胞识别肿瘤细胞,该研究发现 ADAR1 的功能类似于免疫检查点,ADAR1 的缺失恢复了 B2m 缺失的肿瘤对免疫治疗的敏感性,提示:CD8+T 细胞对肿瘤的识别不是对癌细胞有效免疫应答所必需的。
PKR 介导的 dsRNA 应激抑制了肿瘤细胞生长,MDA5 介导的 dsRNA 应激增加了肿瘤炎症,双重机制提高了肿瘤对 IFN 的敏感性,增强了对免疫检查点抑制剂的响应,克服了免疫治疗的抵抗。该研究为克服免疫检查点阻断治疗抵抗提供了新思路、新方法、新策略。
值得注意的是作为核糖核酸腺苷脱氨酶,ADAR1 是否通过其他机制影响肿瘤生物学行为?其在不同肿瘤样本中的表达与突变如何? 该研究尚处于动物试验阶段,在人体试验中的重现性如何?等问题还需要进一步的探索和研究。
缩略词:
ADAR1:adenosine deaminase acting on RNA RNA 核糖核酸腺苷脱氨酶 1
CRISPR:clustered regularly interspersedshort palindromic repeats 成簇的规律间隔的短回文重复序列
dsRNA:double-stranded RNA 双链 RNA
A-to-I: adenosine-to- inosine 腺苷到肌苷
IFN:interferon 干扰素
PD-1:programmed death-1 程序性细胞死亡蛋白-1
NK:natural killer 自然杀伤细胞
MDSC:myeloid-derived suppressor cells 骨髓来源的抑制性细胞
TME :tumour micro-environment 肿瘤微环境
PKR:protein kinase R 蛋白激酶 R
MDA5:Melanoma differentiation associated gene-5 黑色素瘤分化相关基因 5
GM-CSF:granulocyte–macrophage colony-stimulating factor 粒细胞-巨噬细胞集落刺激因子
GVAX:granulocyte–macrophage colony-stimulating factor -secreting whole tumour cell vaccine 粒细胞-巨噬细胞集落刺激因子分泌的全肿瘤细胞疫苗