NDUFA13 —— 心肌保卫者?
11 月 7 日,PNAS 在线发表了浙大医学院朱伟和王建安课题组发表的题为「Electron leak from NDUFA13 within mitochondrial complex I attenuates ischemia-reperfusion injury via dimerized STAT3」的文章,揭示了 NDUFA13 在缺血再灌注损伤过程中对心肌的保护作用。
本研究中 NDUFA13 条件性敲除小鼠由上海南方模式生物构建和繁育。
Highlight
由于线粒体电子泄漏引起的活性氧(ROS)的产生可能参与了很多生理或病理过程。NDUFA13 (NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 13) 是线粒体复合体 I 的辅助亚基,具有独特的分子结构,位于电化学势低的 FeS 簇附近。
研究中建立了心脏特异性 NDUFA13 基因敲除杂合子小鼠。在基础状态下,NDUFA13 的适度下调在复合体 I 内引起电子泄漏,导致轻度增加了细胞质局部的 H2O2。 由此产生的 ROS 作为第二信使,负责 STAT3 的二聚化,因此,激活了抗凋亡信号通路,最终显著抑制超氧化物爆发并减少了缺血再灌注过程中的梗死面积。
小鼠模型
NDUFA13 flox 小鼠:利用经典 ES 细胞打靶技术,将 loxp 位点通过同源重组整合到 NDUFA13 基因 exon3 两侧。该品系由上海南方模式生物构建。
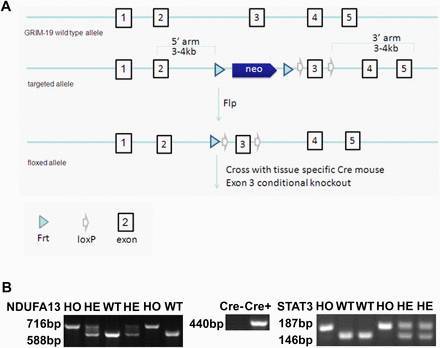
图 1. NDUFA13 flox 小鼠构建策略。
MYH6-CreER 转基因小鼠:可受 Tamoxifen 诱导,在幼年及成年心肌细胞中表达 Cre。
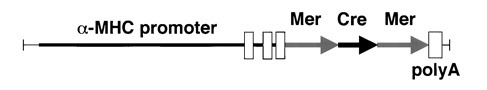
图 2. MYH6-CreER 小鼠构建策略 [1]。
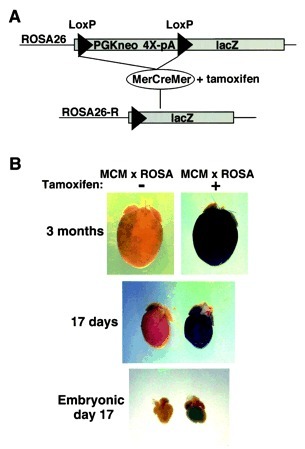
图 3. MYH6-CreER 小鼠与 Rosa26-lacZ 报告基因小鼠交配,验证心肌细胞特异性表达 [1]。
STAT3 flox 小鼠:利用经典 ES 细胞打靶技术,将 loxp 位点通过同源重组整合到 STAT3 基因 exon18-20 两侧。
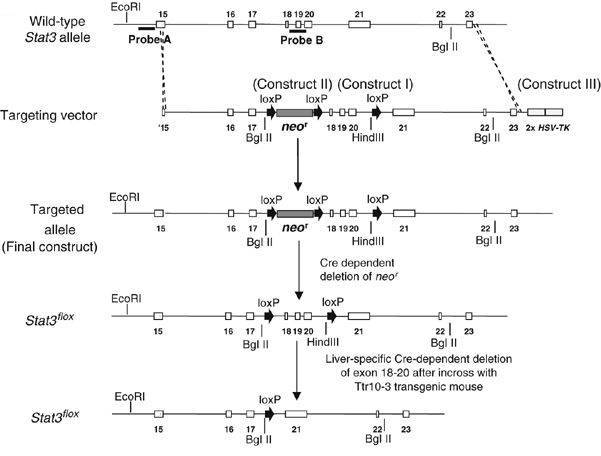
图 4. STAT3 flox 小鼠构建策略 [2]。
研究结果
体外实验表明 NDUFA13 适度下调可防止缺氧/复氧诱导的细胞损伤。
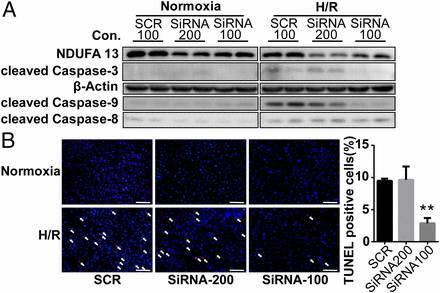
图 5. 在心肌母细胞 H9C2 cells 中对 NDUFA13 基因进行 RNAi, 抑制 NDUFA13 基因表达:100 μmol/L siRNA-NDUFA13 可使表达下降约 30%;200 μmol/L siRNA-NDUFA13 下降约 60%(A)。当缺氧 6 小时后复氧 18 小时时,NDUFA13 中度下调(~30%)的细胞 TUNEL 阳性率显著降低,而 NDUFA13 大幅下调(~60%)则没有减少缺氧/复氧诱导的细胞凋亡(B)。NDUFA13 中度下调时,细胞凋亡内在途径 marker caspase-3 和 caspase-9 也显著下调;而外在途径 caspase-8 则没有变化(A)。
建立 NDUFA13 心肌细胞特异性基因敲除小鼠模型,及初步表型分析。
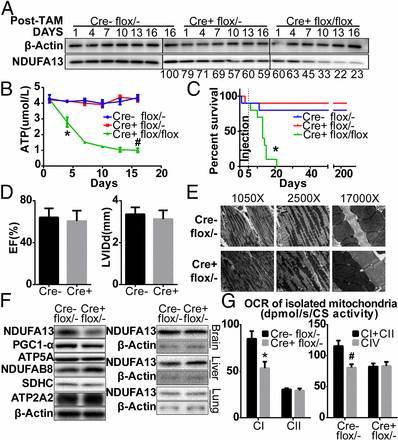
图 6. 8 周龄心肌细胞 NDUFA13 敲除纯合子、杂合子和野生型对照,在给予 Tamoxifen(45 mg/kg, i.p. for 5 days)诱导后第 1、4、7、10、13 以及 16 天分别检测基因敲除效果,发现杂合子小鼠中 NDUFA13 基因在诱导后 16 天被中度下调,而在基因敲除纯合子小鼠中诱导后 1 天就已经达到同等下调水平,诱导后 16 天时 NDUFA13 基因表达已经下降了 80%(A)。不过杂合子小鼠 ATP 水平没有显著变化,存活率与野生型小鼠一致;而纯合子小鼠随着 Tamoxifen 诱导时间的增加,心脏中 ATP 浓度随之降低,从而发生突然的死亡(B-C)。故选择 NDUFA13 敲除杂合子小鼠作为实验组,Tamoxifen 诱导 28 天后,杂合子小鼠与野生型小鼠一样心肌结构与功能、超微结构、线粒体形态都维持正常(D-E);线粒体关键蛋白,如:ATP2A2、NDUFB8、SDHC、ATP5A、PGC-1a,都未受影响(F)。杂合子小鼠心肌细胞线粒体复合物 I 耗氧率(OCR)显著降低,而复合物 I 和 II 的耗氧率补偿性地与复合物 IV 一致(G)。
NDUFA13 下调可保护小鼠在缺血再灌注(I/R)模型中的细胞损伤。
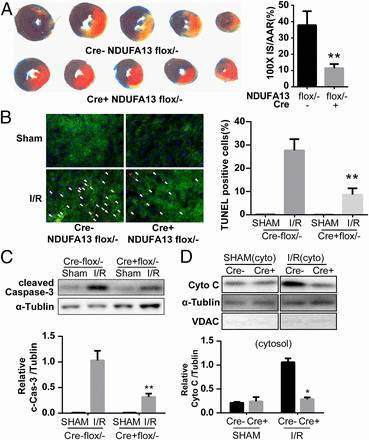
图 7. NDUFA13 心肌细胞特异性敲除杂合子小鼠与野生型对照组均冠状动脉结扎 45 分钟,然后再灌注 3 小时,建立体内缺血再灌注模型(A)。杂合子小鼠 TUNEL 阳性心肌细胞显著减少(B), 梗死周围心脏组织中 cleaved caspase-3 表达也显著降低(C),细胞色素 C 也明显减少(D),说明 NDUFA13 适度下调后在缺血再灌注模型中具有保护细胞减少凋亡的作用。
NDUFA13 下调后增加细胞质 ROS 的生成。
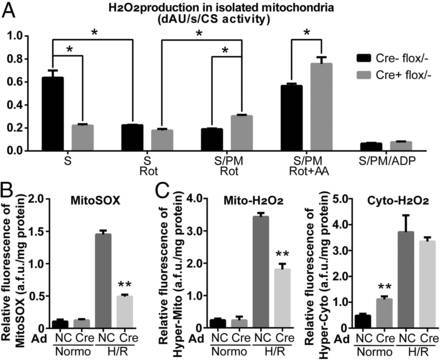
图 8. 用分别针对复合物 I 和复合物 III 的不同底物和抑制剂,检测 NDUFA13 心肌细胞特异性敲除杂合子小鼠线粒体中 OCR 和 H2O2 的水平。结果发现,野生型小鼠的线粒体中琥珀酸会引起反向电子传递(RET)引发的 H2O2 增加,而 NDUFA13 敲除杂合子小鼠中 H2O2 的产生明显减少,说明 NDUFA13 下调导致 RET 受阻。加入丙酮酸和苹果酸后,杂合子小鼠 H2O2 水平上升(A)。NDUFA13 flox 杂合新生小鼠心肌细胞(NMCMs)通过 Cre 表达的腺病毒感染,使 NDUFA13 表达下调。利用 mitoSOX Red 线粒体超氧化物指示剂检测线粒体中超氧化物水平,发现 NDUFA13 敲除和野生型的 NMCMs 中超氧化物水平相似,而缺氧/复氧诱导处理后 NDUFA13 敲除的 NMCMs 中超氧化物水平显著下降(B)。利用 HyPer H2O2 探针分别检测线粒体与细胞质中 H2O2 的水平,发现正常状态下,NDUFA13 敲除的 NMCMs 细胞质中 H2O2 水平上升,线粒体中没有变化;缺氧/复氧诱导处理后,野生型 NMCMs 中 H2O2 爆发,而 NDUFA13 敲除的 NMCMs 中 H2O2 水平上升幅度明显降低(C)。
NDUFA13 的 TMH 结构维持线粒体膜完整性。
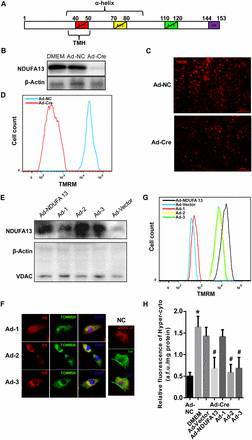
图 9. 在 NDUFA13 敲除的 NMCMs 中过表达破坏不同结构域的 NDUFA13 突变体(A):Ad-1 (with a deletion of amino acid 40–50), Ad-2 (a deletion of amino acids 70–80), Ad-3 (a deletion of amino acids 110–120), Ad-NDUFA13 (a wild-type full-length NDUFA13 as a normal control), and Ad-Vector (an empty vector as a negative control),结果发现破坏了 TMH 结构域的 NDUFA13 突变体(Ad-1)不能定位到线粒体以维持线粒体膜电位(F-G),也不能消除细胞质中 H2O2 水平上升(H),而 Ad-2 和 Ad-3 则与野生型 NDUFA13 功能一致,说明 NDUFA13 的 TMH 结构域对于维持 MMP 非常重要,同时也是 H2O2 产生的主要来源。
适度的 NDUFA13 下调通过 STAT3 行使保护心肌的作用。
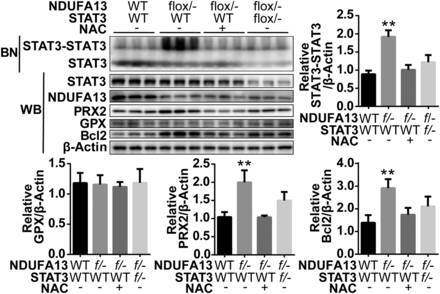
图 10.NB-PAGE 检测当给予 NAC(ROS 清道夫)或 STAT3 敲低后(将 NDUFA13 flox 小鼠与 STAT3 flox 小鼠交配,获得双杂合小鼠),STAT3 低聚物以及 GPX、PRX2、Bcl2 的表达情况。NDUFA13 下调引起 PRX2 表达上调,会增加 STAT3 二聚化,从而上调 Bcl-2 表达;当 ROS 被清除后,PRX2 的上调效应消失。
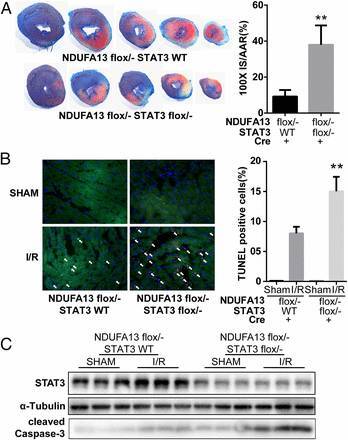
图 11. 心肌特异性 NDUFA13 敲除杂合子小鼠和心肌特异性 NDUFA13/STAT3 敲除双杂合子小鼠给予 Tamoxifen 诱导两周后进行缺血再灌注造模。STAT3 敲低后,NDUFA13 下调的保护作用消失了,梗死面积增加(A),梗死周围 TUNEL 阳性细胞增加(B),cleaved caspase-3 表达上调(C)。
NDUFA13 作用机制
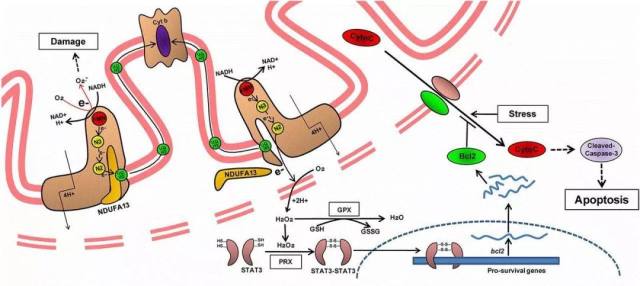
图 12. 在野生型小鼠中(上图左侧),通常缺血再灌注损伤后会导致超氧化物的显著增加,导致细胞损伤。在 NDUFA13 适度敲低的小鼠中(上图右侧),会产生一部分电子泄漏。由于 NDUFA13 位于复合体 I 的低电化学势位置,一小部分过氧化氢在基础状态下产生,反过来激活了 PRX2,导致 STAT3 二聚化,上调 Bcl-2 抑制细胞凋亡。
